
本指南将从肥厚型心肌病(HCM)的定义与流行病学,病因和病理生理机制,临床评估,辅助检查,诊断标准及流程,临床分型及分期,并发症,遗传评估和家系筛查,鉴别诊断,治疗,心脏性猝死的风险评估、预防和治疗,特殊人群管理,自然病程及预后等方面详细介绍HCM的诊断、治疗和管理,目的是在国内推广关于HCM的最新诊疗成果,进一步提高我国HCM的诊疗水平。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
肥厚型心肌病(hypertrophic cardiomyopathy,HCM)是一种最常见的遗传性心脏病[1, 2, 3, 4]。对HCM的现代研究始于20世纪60年代[5, 6, 7, 8],经过70年的发展,如今对于HCM的病因、诊断、治疗和管理等方面均取得了巨大进展[9, 10]。
2017年,由中国医师协会心力衰竭专业委员会组织国内HCM相关领域的专家撰写并发表了《中国肥厚型心肌病管理指南2017》(简称2017年指南)[2],促进了我国HCM的规范化诊治和管理。近5年来,在HCM的诊断和治疗方面又有了一些新的进展。为在国内推广关于HCM的最新诊疗成果,进一步提高我国HCM的诊疗水平,由国家心血管病专家委员会心力衰竭专业委员会、中国医师协会心力衰竭专业委员会、中华医学会心血管分会心力衰竭学组联合中华心力衰竭和心肌病杂志编辑委员会,组织国内从事HCM相关领域研究的专家,通过分析总结目前国内外最新研究证据,撰写了《中国肥厚型心肌病指南2022》。
本指南采用国际通用方式对诊疗措施标明相应的推荐类别(表1)和证据水平(表2)。

推荐类别分类、定义及相关术语
推荐类别分类、定义及相关术语
| 推荐级别 | 定义 | 术语 |
|---|---|---|
| Ⅰ类 | 指有证据已证实和(或)一致公认有益、有用和有效的操作或治疗 | 推荐/建议 |
| Ⅱ类 | 指有用和(或)有效的证据尚有矛盾或存在不同观点的操作或治疗 | |
| Ⅱa类 | 指有关的证据/观点倾向于有用和(或)有效,应用这些操作或治疗是合理的 | 应该考虑 |
| Ⅱb类 | 指有关的证据/观点尚不能被充分证明有用和(或)有效,应用这些操作或治疗可能是合理的 | 可以考虑 |
| Ⅲ类 | 指有证据已经证实和(或)一致公认为无用和(或)无效,甚至对一些病例可能有害的操作或治疗 | 不推荐/不建议 |

证据水平分类及定义
证据水平分类及定义
| 证据水平 | 定义 |
|---|---|
| 证据水平A | 指资料来源于多项随机对照研究或荟萃分析 |
| 证据水平B | 指资料来源于单项随机对照研究或多项非随机对照研究 |
| 证据水平C | 指仅为专家共识、建议、小型临床研究、回顾性研究或注册登记研究 |
HCM是一种呈常染色体显性遗传的原发性心肌病。主要由编码心肌肌小节相关蛋白的基因致病性变异引起,临床表现以心室壁增厚为突出特征。需除外其他可引起心室壁增厚的生理因素、心脏疾病、系统性疾病或代谢性疾病。
HCM是一种全球性的疾病,早期流行病学调查显示,普通成人HCM患病率为0.16%~0.23%[11, 12, 13, 14],平均为0.20%(1/500)。之后研究发现,普通人群携带致病性肌小节基因变异的发生率高于既往预期[15];基因检测技术的进步,发现了更多携带致病性肌小节基因变异但临床无左心室肥厚(left ventricular hypertrophy,LVH)表现的患者,称为“基因型阳性表型阴性”个体[16];心脏磁共振(cardiac magnetic resonance,CMR)检查等现代心脏影像学技术的发展,有助于识别更多既往超声心动图检查不能识别或容易漏诊的HCM表型[17]。因此,估测临床表达的HCM和未表达的基因携带者的患病率高于既往调查结果,可以达到1/200(0.5%)[18]。
1. 致病基因:目前认为,多数HCM是一种单基因遗传性心脏病,大约60%的HCM患者存在致病基因变异,主要编码心肌肌小节相关蛋白,包括粗肌丝、中间丝及细肌丝等[1, 2, 3, 4,9, 10]。自从1990年Geisterfer-Lowrance等人[19]首次在HCM家系中发现编码心肌肌小节粗肌丝的β-肌球蛋白重链(myosin heavy chain 7,MYH7)基因是致病基因以来,迄今已在至少8个编码心肌肌小节相关蛋白的基因(被称为HCM的“核心致病基因”)中发现>1 500个与HCM发病相关的变异(表3)[10],其中,心脏型肌球 蛋白结合蛋白C(cardiac myosin binding protein C3,MYBPC3)基因和MYH7基因是HCM患者最常见的两种致病基因,二者约占基因变异阳性患者的70%。
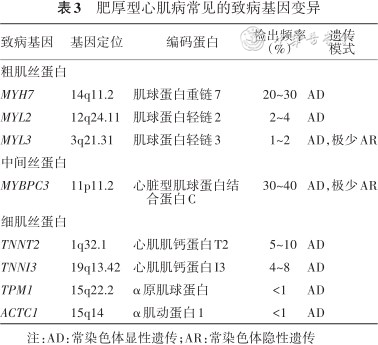
肥厚型心肌病常见的致病基因变异
肥厚型心肌病常见的致病基因变异
| 致病基因 | 基因定位 | 编码蛋白 | 检出频率(%) | 遗传模式 |
|---|---|---|---|---|
| 粗肌丝蛋白 | ||||
| MYH7 | 14q11.2 | 肌球蛋白重链7 | 20~30 | AD |
| MYL2 | 12q24.11 | 肌球蛋白轻链2 | 2~4 | AD |
| MYL3 | 3q21.31 | 肌球蛋白轻链3 | 1~2 | AD,极少AR |
| 中间丝蛋白 | ||||
| MYBPC3 | 11p11.2 | 心脏型肌球蛋白结合蛋白C | 30~40 | AD,极少AR |
| 细肌丝蛋白 | ||||
| TNNT2 | 1q32.1 | 心肌肌钙蛋白T2 | 5~10 | AD |
| TNNI3 | 19q13.42 | 心肌肌钙蛋白I3 | 4~8 | AD |
| TPM1 | 15q22.2 | α原肌球蛋白 | <1 | AD |
| ACTC1 | 15q14 | α肌动蛋白1 | <1 | AD |
注:AD:常染色体显性遗传;AR:常染色体隐性遗传
2. 遗传模式:多数由基因变异引起的HCM的遗传模式为常染色体显性遗传模式,每个受累家系成员的后代都有50%的概率会遗传到这种致病变异。但由于致病基因变异存在外显不全及与年龄依赖的表达,携带致病基因变异不一定出现临床表型[10]。
3. 复杂基因变异:部分HCM患者可能携带单个基因的多个变异(复合变异)或≥2个相同或不同基因的杂合变异,统称为复杂基因变异现象。研究发现,携带复杂基因变异者发病更早,临床表型更重,预后更差[20]。
4. 基因检测阴性病例
目前研究发现,仍有高达40%的HCM患者未检测到致病基因变异,主要见于一些散发病例(称为“非家族性HCM”)或小型家系,通常发病较晚,临床表型相对较轻[21],提示可能有不同机制参与HCM发病。
HCM具有复杂的病理生理机制,主要包括左心室流出道梗阻(left ventricular outflow tract obstruction,LVOTO)、二尖瓣反流(mitral regurgitation,MR)、舒张功能不全、心肌缺血和自主神经功能不全等[1, 2, 3, 4]。对于一个具体的HCM患者,可能以一种机制为主,也可能涉及多种机制之间复杂的相互作用。
1. LVOTO:(1)诊断标准:目前,LVOTO主要采用多普勒超声心动图检查来评估,定义为左心室流出道(left ventricular outflow tract,LVOT)瞬时峰值压差≥30 mmHg(1 mmHg=0.133 kPa)。(2)产生机制:目前认为,主要有两种机制参与LVOTO的产生:①非对称性室间隔肥厚(asymmetric septal hypertrophy,ASH),尤其是室间隔基底部增厚及二尖瓣解剖结构改变(包括瓣叶拉长、乳头肌前移)造成LVOT狭窄引起的机械性梗阻;②二尖瓣前叶收缩期前移(systolic anterior motion,SAM)现象加重LVOT狭窄引起的动力性梗阻[22]。(3)影响因素:HCM患者LVOTO是动态变化的,对心室负荷及心肌收缩力变化敏感[23]。增强心肌收缩力或减轻心脏前后负荷(如使用强心药、采取站立位、Valsalva动作、含服硝酸甘油、使用血管扩张剂等)可使LVOTO加重;相反,减弱心肌收缩力或增加心脏前后负荷(如使用β受体阻滞剂、采取蹲位、抬腿等)可使LVOTO减轻。(4)激发试验:对静息状态不存在LVOTO的HCM患者,需进行激发试验[24, 25]结合超声心动图检查,评估是否存在隐匿性LVOTO。激发试验可以采用Valsalva动作,或床旁重复仰卧-站立位或蹲下-站立位,直立位踏车试验或半卧位自行车试验等。由于缺乏特异性,目前指南不再推荐使用多巴酚丁胺药物负荷试验[1, 2, 3, 4]。(5)预后影响:LVOTO增加左心室收缩压,加重心肌缺血,是梗阻性HCM患者症状产生的主要原因。研究显示,LVOTO显著增加了HCM患者发生心脏性猝死(sudden cardiac death,SCD)、进展至中重度心力衰竭(心衰)、卒中及死亡等不良事件的风险[26, 27]。
2. MR:产生原因主要有两方面:(1)继发于LVOTO导致的SAM现象;(2)原发性或内在性的二尖瓣相关结构异常,如二尖瓣叶过度拉长、异常乳头肌插入、乳头肌前向移位等[28]。
3. 舒张功能不全:产生机制主要包括两方面:(1)心肌缺血、缺氧、能量代谢障碍,导致心肌细胞内舒张期钙再摄取异常,导致心肌主动松弛能力受损;(2)心室肥厚、心肌纤维化、心室几何形状改变等,导致心室壁顺应性降低(僵硬度增加),心室被动充盈受限[1, 2, 3, 4]。对于非梗阻性HCM患者,舒张功能不全是导致活动耐力下降,出现心衰症状的主要原因。由于心室松弛功能受损,心室充盈更依赖于心房收缩,当出现心房颤动(房颤)等房性心律失常时,HCM患者的耐受性更差。
4. 心肌缺血:产生原因可能包括如下几个方面:(1)心肌细胞肥大,导致心肌的氧供和需求失衡;(2)心室壁内小冠状动脉管壁增厚,管腔狭窄,血管分布密度降低[29];(3)冠状动脉微血管功能障碍(microvascular dysfunction,MVD)导致冠状动脉血流储备降低;(4)部分患者可以合并冠状动脉肌桥及冠状动脉粥样硬化病变。
5. 自主神经功能异常:主要表现为心率恢复异常[30]、不恰当的血管扩张和对运动的异常血压反应(abnormal blood pressure response,ABPR)[31],与HCM的预后有一定关系。
HCM患者的临床评估包括家族史、症状和体征等。对于可疑HCM患者,首次评估时应进行全面的体格检查,详细的个人史采集及至少3代亲属的家族史采集(Ⅰ类推荐,B级证据)。
家族史对于HCM患者的诊断及预后评估非常重要。有意义的家族史包括家族成员诊断HCM或一级亲属在年龄≤50岁发生SCD、心衰、心脏移植及植入式心脏转复除颤起搏器(implantable cardioverter defibrillator,ICD)治疗史等[32]。
HCM的临床表现差异较大,多数HCM患者临床无症状,是在体格检查或因其他疾病行心电图或超声心动图检查时意外发现;部分患者可以有运动相关的症状,如呼吸困难、胸痛、心悸及晕厥等;部分患者以SCD为首发表现。
1. 呼吸困难:部分患者可出现劳力性呼吸困难,与LVOTO及心室舒张功能异常相关。
2. 胸痛:部分患者有劳力性胸痛症状,与心肌缺血相关。
3. 心悸:患者可出现多种心律失常,包括房性和室性心律失常。
4. 晕厥或晕厥前状态:研究显示,约15%~25%的HCM患者至少发生过1次晕厥或晕厥前状态[33]。原因主要为心律失常和(或)血流动力学异常(LVOTO和自主神经功能异常)。研究显示,首次评估前6个月内发生不明原因晕厥是HCM患者发生SCD的独立预测因素,其风险是无晕厥患者的近5倍,这种关系在不同年龄组中均存在[34]。
5. SCD:主要机制是致命性室性心律失常,也可能是血流动力学异常。
梗阻性HCM患者的体征主要与LVOTO有关。位于胸骨左缘第3~4肋间可以闻及较粗糙的喷射性收缩期杂音,不向颈部传导,增强心肌收缩力或减轻心脏前后负荷可使杂音增强;相反,减弱心肌收缩力或增加心脏前后负荷措施可以使杂音减弱。这种杂音需要与MR以及主动脉瓣狭窄(aortic stenosis,AS)的杂音(下文介绍)进行鉴别。
HCM患者在初次评估及定期随访过程中需要完善相关辅助检查,包括以下几个方面[35]。
主要包括常规12导联心电图和24~48h动态心电图检查。
1. 常规12导联心电图检查:可以提供关于左心室高电压、复极异常及心律失常等信息[36]。一项研究显示,HCM患者首次就诊时94%存在心电图异常,包括左心室高电压、病理性Q波及ST-T改变等,只有6%患者的心电图正常[37]。常规12导联心电图检查可以用于HCM患者的初始评估、定期随访及家系成员的筛查[36]。对于诊断HCM患者,初始评估和后续定期随访(每1~2年)推荐进行常规12导联心电图检查(Ⅰ类推荐,B级证据);对于诊断HCM患者的一级亲属,推荐开展常规12导联心电图检查进行家系筛查(Ⅰ类推荐,B级证据)。
2. 24~48h动态心电图检查:可见不同类型的心律失常,包括室上性和室性心律失常[38]。其中,非持续性室性心动过速(non-sustained ventricular tachycardia,NSVT,定义为≥3个连续室性心搏,心率≥100 次/min,持续时间<30s)发生率在20%~30%之间[39],与年龄<35岁的年轻患者SCD风险增加独立相关;而快速心室率(>200次/分)、持续更长(>7阵)及反复发作NSVT对于ICD治疗的室性心律失常的预测价值更大[40]。对于诊断HCM患者,初始评估和后续定期随访(每1~2年)推荐进行24~48h动态心电图监测(Ⅰ类推荐,B级证据)。对于诊断HCM患者,如果出现心悸或头痛症状,推荐进行延长(>24h)心电监测及事件记录(Ⅰ类推荐,B级证据)。
超声心动图检查是目前HCM患者临床诊断、病情监测、治疗方法选择及治疗效果评价首选检查方法。目前,有多种超声检查新技术应用于临床,如组织多普勒成像(tissue Doppler imaging,TDI)、心肌超声声学造影(myocardial contrast echocardiography,MCE)、斑点追踪超声心动图(speckle tracking echocardiography,STE)进行心肌应变/应变率成像等,其应用主要包括以下几个方面[41, 42, 43]。
1. 测量心室壁厚度,评估LVH:需要测量舒张末期心室短轴从心底至心尖所有节段的最大心室壁厚度,避免心室长轴M型超声斜切而高估心室壁厚度。LVH一般采用左心室质量指数(left ventricular mass index,LVMI)来评价。
2. 测量LVOT压差,评估LVOTO:首先测量静息状态下LVOT的瞬时峰值压差,评估是否存在静息性LVOTO;如果静息状态下LVOT压差<50 mmHg,推荐进行激发试验,评估是否存在隐匿性LVOTO(Ⅰ类推荐,B级证据)。
3. 其他:评估二尖瓣功能或结构异常,评估左心室舒张功能异常等。
MCE检查:用于常规经胸超声心动图(transthoracic echocardiography,TTE)检查不能明确诊断的心尖部肥厚或者心尖部室壁瘤、血栓形成(Ⅱa类推荐,B级证据),特别对于计划行酒精间隔消融术(alcohol septal ablation,ASA)的HCM患者可经冠状动脉内注射心肌声学造影剂进行MCE检查(Ⅰ类推荐,B级证据)。
经食道超声心动图(transesophageal echocardiography,TEE)检查:对于常规TTE检查不能明确的HCM患者,应该考虑进行TEE检查协助评估LVOTO机制及二尖瓣病变情况,指导治疗决策(Ⅱa类推荐,C级证据)。对于行外科室间隔切除术(surgical septal myectomy,SSM)的患者,推荐术中TEE检查,有助于准确评估LVOTO机制及二尖瓣病变情况,指导手术策略,并监测术后并发症(Ⅰ类推荐,B级证据)。
4. 主要推荐意见:对于可疑HCM患者,初始评估推荐进行常规TTE检查(Ⅰ类推荐,B级证据)。对于诊断HCM患者,如果临床状况稳定,推荐每1~2年复查常规TTE,评估心肌肥厚程度、LVOTO、MR及心功能水平(Ⅰ类推荐,B级证据);如果临床状况发生变化或出现新的临床事件,推荐及时复查常规TTE(Ⅰ类推荐,B级证据)。已经行室间隔减容术(septal reduction therapy,SRT)的HCM患者,推荐术后3~6个月复查常规TTE,评估治疗效果(Ⅰ类推荐,B级证据)。对于诊断HCM患者的一级亲属,推荐初始家系筛查及定期随访时进行常规TTE检查(Ⅰ类推荐,B级证据);针对HCM家系中“基因型阳性表型阴性”个体,推荐儿童和青少年每1~2年,成人每3~5年复查一次常规TTE(Ⅰ类推荐,B级证据)。
CMR检查是目前诊断HCM最准确的检查方法,也是目前评估心肌纤维化首选的无创性影像学方法[35,44],在HCM患者中的应用主要包括以下几个方面。
1. 测量心室壁厚度及心腔大小:对于左心室前侧壁、后间隔、右心室、心尖部等特殊部位肥厚或心尖部室壁瘤、血栓形成,CMR检查要明显优于常规TTE检查,有助于HCM的准确分型。
2. 评估心肌纤维化:LGE可以评估心肌局限的替代性纤维化(心肌瘢痕),有助于HCM的危险分层及预后判断[45, 46]。研究显示,存在广泛延迟钆强化(late gadolinium enhancement,LGE)(如≥15%左心室质量)显著增加HCM患者的SCD风险[45],与HCM患者不良预后相关。
基因检测在HCM的诊断和鉴别诊断、家系筛查及优生优育等方面均具有重要的价值[47, 48],在HCM的预后评价和危险分层方面也具有一定价值[49, 50]。对于诊断HCM的患者,推荐进行基因检测,明确遗传基础,有助于识别家系中发生HCM的高危个体(Ⅰ类推荐,B级证据)。
由于HCM主要是由编码心肌肌小节相关蛋白的基因变异引起,基因变异形式主要是错义突变。目前主要采用二代测序(next generation sequencing,NGS)技术进行基因突变检测,具有高通量、检测快、成本低等特点[51]。
应用NGS技术进行基因检测时,通常首选包含8个与HCM发病明确相关的“核心致病基因”(MYH7、MYBPC3、MYL2、MYL3、TNNT2、TNNI3、TPM1和ACTC1基因)的“靶基因”或“基因组合(gene panels)”测序(Ⅰ类推荐,B级证据)。如果上述基因检测未能明确致病基因突变,可以考虑扩展为针对心肌病的基因组合测序,或全外显子组测序或全基因组测序[52]。如果考虑其他HCM“拟表型”时,推荐进行相关特定基因突变检测(Ⅰ类推荐,B级证据)。基因变异检测结果只有致病(pathogenic,P)和很可能致病(likely pathogenic,LP)类型的变异才是有临床意义的[53]。
需要指出的是,基因变异的致病性依赖于当前的证据数量或水平,随着时间变化,基因变异的致病性分类可能发生变化。因此,对于已经进行基因检测的HCM患者,推荐定期对基因变异的致病性进行再评价,判定是否需要再分类(Ⅰ类推荐,B级证据)[54]。
HCM患者部分合并冠状动脉肌桥或冠状动脉粥样硬化性心脏病(冠心病),与不良预后相关[55]。其主要应用包括如下几个方面。
1. 对于有心绞痛症状或心肌缺血证据的HCM患者,推荐进行冠状动脉造影或CT检查以明确冠心病诊断并指导治疗决策(Ⅰ类推荐,B级证据)。
2. 有冠心病危险因素的HCM患者计划行外科室间隔切除术的患者,术前推荐进行冠状动脉造影检查,评估是否合并冠心病及其程度,指导治疗决策(Ⅰ类推荐,B级证据)。
3. 对于计划进行酒精室间隔消融术的HCM患者,术前推荐进行冠状动脉造影检查,评估是否合并冠心病及其程度,同时了解间隔支动脉情况,指导治疗决策(Ⅰ类推荐,B级证据)。
4. 心室造影可以显示左心室在收缩期及舒张期的形态改变,有助于识别不同类型HCM患者;可以连续测量左心室腔与升主动脉之间的压差,有助于梗阻性HCM与AS的鉴别诊断。
主要包括平板运动试验和心肺运动试验(cardiopulmonary exercise test,CPET)。在HCM患者中的应用如下:
1. 平板运动试验:可以评估HCM患者对运动的ABPR,通常定义为运动时收缩压不能升高>20 mmHg或较运动高峰时收缩压下降>20 mmHg,20%~40%成人HCM患者存在ABPR,与年龄<50岁HCM患者的SCD风险增高相关[56]。
2. CPET:可以客观评价HCM患者的心肺功能,对活动耐力或功能受限程度进行定量评估,与HCM患者的不良预后相关[57]。对于终末期(end stage,ES)HCM(ES-HCM)患者,可以协助心脏移植或机械循环支持(mechanical circulatory support,MCS)适应证的评估。
目前主要指利钠肽检测及心肌肌钙蛋白检测。利钠肽检测用于评估心脏负荷情况或心脏功能状态,心肌肌钙蛋白检测用于评估心肌损伤情况,二者均有助于HCM患者危险分层及预后判断[57, 58, 59, 60, 61]。因此,对于HCM患者初始评估及定期随访过程中,推荐常规检测上述心脏生物标志物指标(Ⅰ类推荐,C级证据)。
主要指心导管检查,适用于以下两种情况:
1. 诊断HCM患者计划行SRT,但术前无创性影像学检查不能准确评估LVOTO程度,推荐进行心导管有创性血流动力学评估,指导治疗决策(Ⅰ类推荐,B级证据)。
2. 诊断ES-HCM患者计划进行心脏移植或MCS,术前常规进行心导管有创性血流动力学评估(Ⅰ类推荐,C级证据)。
心内电生理检查(electrophysiological examination,EPS)及导管消融治疗主要用于以下两种情况[1]。
1. 证实有持续的或反复发作的室上性心动过速及心室预激的HCM患者,推荐进行EPS及导管消融治疗(Ⅰ类推荐,C级证据)。
2. 部分明确的、症状性、持续性单形室性心动过速患者,可以考虑进行EPS及导管消融治疗(Ⅱb类推荐,C级证据)。
心内膜心肌活检(endocardial myocardial biopsy,EMB)属于有创性检查项目,有一定的并发症风险,限制了其临床广泛应用。
目前,EMB主要用于临床评估提示浸润性、贮积性或炎症性疾病(HCM的“拟表型”)而常规无创性影像学检查无法明确诊断,或者HCM患者经常规治疗后效果不佳需要进一步明确病因的情况下,有助于明确诊断,指导治疗[62]。
心室壁增厚是诊断HCM的必备条件。可以利用不同的心脏影像学检查方法如超声心动图、CMR或心脏CT成像检查等。
1. 成人(年龄≥18岁)HCM的诊断标准:(1)上述任一心脏影像学检查发现一个或多个左心室节段舒张末期最大心室壁厚度≥15 mm。其中,左心室壁最大厚度≥30 mm称为极度左心室肥厚[63]。(2)对于家族性HCM中除先证者外的家庭成员或基因检测阳性(携带HCM致病基因变异)的个体,舒张末期最大心室壁厚度≥13 mm也可以诊断HCM。
2. 儿童(年龄<18岁)HCM的诊断标准:根据儿童年龄、体表面积(m2)、筛查环境及诊断HCM的验前概率,采用不同的诊断界值:(1)对于无HCM家族史且无症状的儿童,当左心室壁最大厚度超过预测正常值的2.5个标准差,即z值(定义为偏离同年龄儿童正常值的标准差数)>2.5时可诊断HCM。(2)对于有明确HCM家族史或者致病基因检测阳性的儿童,建议采用z值>2的界值。
根据血流动力学、肥厚部位及遗传学规律,HCM可以有不同的临床分型。
1. 根据血流动力学分为:(1)梗阻性HCM:根据梗阻部位又可以分为LVOTO、左心室中部梗阻及左心室心尖部梗阻,与心室壁肥厚部位有关。通常所说的梗阻性HCM是指LVOTO,即LVOT瞬时峰值压差≥30 mmHg;可以分为静息梗阻性(静息状态存在LVOTO)和隐匿梗阻性(静息无梗阻,激发试验时出现LVOTO)。(2)非梗阻性HCM:指静息时和激发时LVOT峰值压差均<30 mmHg。依据血流动力学分型是临床最常用的HCM分型方法,有利于指导治疗措施选择。临床上,静息梗阻性、隐匿梗阻性和非梗阻性HCM三种类型约各占1/3[64]。
2. 根据肥厚部位分为:(1)室间隔肥厚:是临床最常见的表现型,主要累及心室间隔基底部,表现为ASH。诊断标准为舒张末期室间隔与左心室后壁厚度之比≥1.3~1.5。(2)心尖部肥厚:又称心尖肥厚型心肌病(apical hypertrophic cardiomyopathy,ApHCM),指心室肥厚主要累及左心室乳头肌以下的心尖部[65]。诊断标准为舒张末期左心室心尖部最大室壁厚度≥15 mm,左心室心尖部与后壁最大厚度之比≥1.5。心电图典型特征是巨大负相T波(giant negative T wave,GNT),电压常>1.0 mV,可见于Ⅰ、aVL、V3-6导联,伴相应导联R波增高及ST段压低,以V4导联最为显著。(3)左心室中部肥厚:又称心室中部梗阻性肥厚型心肌病(mid-ventricular obstructive hypertrophic cardiomyopathy,MVOHCM),是指左心室中部乳头肌水平及心室间隔中部心肌肥厚,伴左心室心尖部与基底部之间收缩末期压差[66]。诊断标准包括:①显著的左心室中部室壁增厚,舒张末期最大室壁厚度≥15 mm,有明确家族史的室壁厚度≥13 mm也可诊断;②左心室中部收缩末期峰值压差≥30 mmHg,常伴有特征性收缩末期异常高速血流(由心尖至心底部)。
3. 根据遗传学特点分为:(1)家族性/遗传性HCM:发病呈家族聚集性,由致病基因变异遗传引起,其诊断标准为除先证者以外,3代亲属中有≥2个成员被诊断为HCM或者存在与先证者相同的基因变异,伴或不伴有心电图及超声心动图异常。(2)散发性HCM:发病无家族性聚集,非基因变异引起,或者患者携带的变异为“原始变异”。
1. 临床分期:2012年,Olivotto等人[67]基于临床和疾病进展的客观证据,提出了一种HCM患者临床分期,共分为4期:Ⅰ期(nonhypertrophic,无肥厚期/临床前期)、Ⅱ期(classic phenotype,典型表型期)、Ⅲ期(adverse remodeling,不良重构期)和Ⅳ期(overt dysfunction,显著功能障碍期/终末期)(表4)。

基于临床和疾病进展客观证据的肥厚型心肌病患者分期
基于临床和疾病进展客观证据的肥厚型心肌病患者分期
| 分期 | 占HCM的比例 | LVEF | LGE(占整个左室质量比) | 冠状动脉MVD | 症状和功能受限 | 左心室充盈异常和TDI | LVOTO(静息或激发) |
|---|---|---|---|---|---|---|---|
| Ⅰ期 | - | 正常或高于正常 | 不存在 | 不详,可能存在 | 无 | 正常,TDI-e′可能降低 | 不存在 |
| Ⅱ期 | ~75% | >65% | 不存在或<5% | 轻度至重度不等 | 不等,可能严重 | 正常或松弛延迟,TDI-e′通常降低 | 常见(70%) |
| Ⅲ期 | ~15% | 50%~65% | 10%~15% | 中度至重度 | 不等,通常轻度至中度 | 假性正常或限制性TDI-e′降低 | 不常见,先前梗阻可能消失 |
| Ⅳ期 | 5%~10% | <50% | 广泛(25%~50%) | 重度 | 通常中度至重度 | 假性正常或限制性TDI-e′严重降低 | 不存在,先前梗阻可能消失 |
注:HCM:肥厚型心肌病,LVEF:左心室射血分数,LVOTO:左心室流出道梗阻;LGE:钆延迟强化,MVD:微血管功能异常,TDI:组织多普勒成像
2. ES-HCM:(1)定义及诊断标准:HCM患者出现严重左心室收缩功能障碍称为ES-HCM,主要诊断标准是超声心动图检查左心室射血分数(left ventricular ejection fraction,LVEF)<50%[68]。ES-HCM患者可以伴有左心室扩大,即左心室舒张末期内径(left ventricular end-diastolic diameter,LVEDD)≥55 mm[68]或不伴有左心室扩大(LVEDD<55 mm)[68]。(2)发生率:目前文献报道,ES-HCM患者占HCM的比例为2.4%~15.7%[68, 69, 70, 71, 72, 73],每年有0.5%~1.5%的HCM患者进展至ES-HCM[68, 69, 70, 71]。(3)疾病预后:早期研究显示,ES-HCM患者的临床表型较重,预后差,年病死率为11.0% [68],心衰和SCD为主要死因。但新近Rowin等人[73]研究显示,在当前抗心衰治疗情况下,ES-HCM患者的年病死率显著下降至1.9%,明显低于既往报道的年病死率(8.0%),但仍高于LVEF保留HCM患者的年病死率(0.2%)。提示,包括ICD和心脏移植等在内的当前治疗策略,显著改善了ES-HCM患者的预后。
3. HCM伴有限制型表型(HCM with restrictive phenotype,RP-HCM):(1)诊断标准:根据Kubo等人[74]的研究,RP-HCM超声心动图诊断标准包括以下几点:①双心房明显扩大,二尖瓣血流E/A比值≥2和减速时间≤150 ms,房颤患者满足后者即可;②不存在或仅轻度LVH;③心室腔缩小或大小正常;④LVEF正常或轻度降低。(2)发生率:不同研究中RP-HCM比率在1.5%~5.9%,提示RP-HCM并不少见[74, 75]。研究显示,RP-HCM患者携带MYH7基因、TNNI3基因和MYL2基因变异的比例较高[74,76]。(3)疾病预后:与典型HCM患者比较,RP-HCM患者临床表型重,预后差,心衰病死率高[74,76]。
房颤是HCM患者最常见的心律失常之一。约18%~20%的HCM患者合并房颤[77, 78, 79],其年发生率约为3%[78]。高龄、左心房增大、纽约心脏协会(New York Heart Association,NYHA)心功能分级Ⅲ级和Ⅳ级是HCM患者发生房颤的主要危险因素。存在上述危险因素的HCM患者在初始评估及定期(每1~2年)随访过程中应该考虑延长(>48h)的动态心电监测评估是否合并房颤(Ⅱa类推荐,B级证据)。房颤影响HCM患者的生活质量,增加患者卒中和周围栓塞的风险,其患病率和年发生率分别为27%和4%[78]。研究显示,经过当前有效治疗,房颤相关病死率降低,已经与无房颤的HCM患者无明显差异,但房颤仍是HCM患者血栓事件的重要病因[79]。
HCM患者可以出现呼吸困难症状,主要由左心室舒张功能异常引起,表现为射血分数保留的心力衰竭(heart failure with preserved ejection fraction,HFpEF,LVEF≥50%)。少数患者进展至ES-HCM,表现为射血分数降低的心力衰竭(heart failure with reduced ejection fraction,HFrEF,LVEF≤40%)和射血分数轻度降低的心力衰竭(heart failure with mildly reduced ejection fraction,HFmrEF,LVEF 41%~49%)[80]。
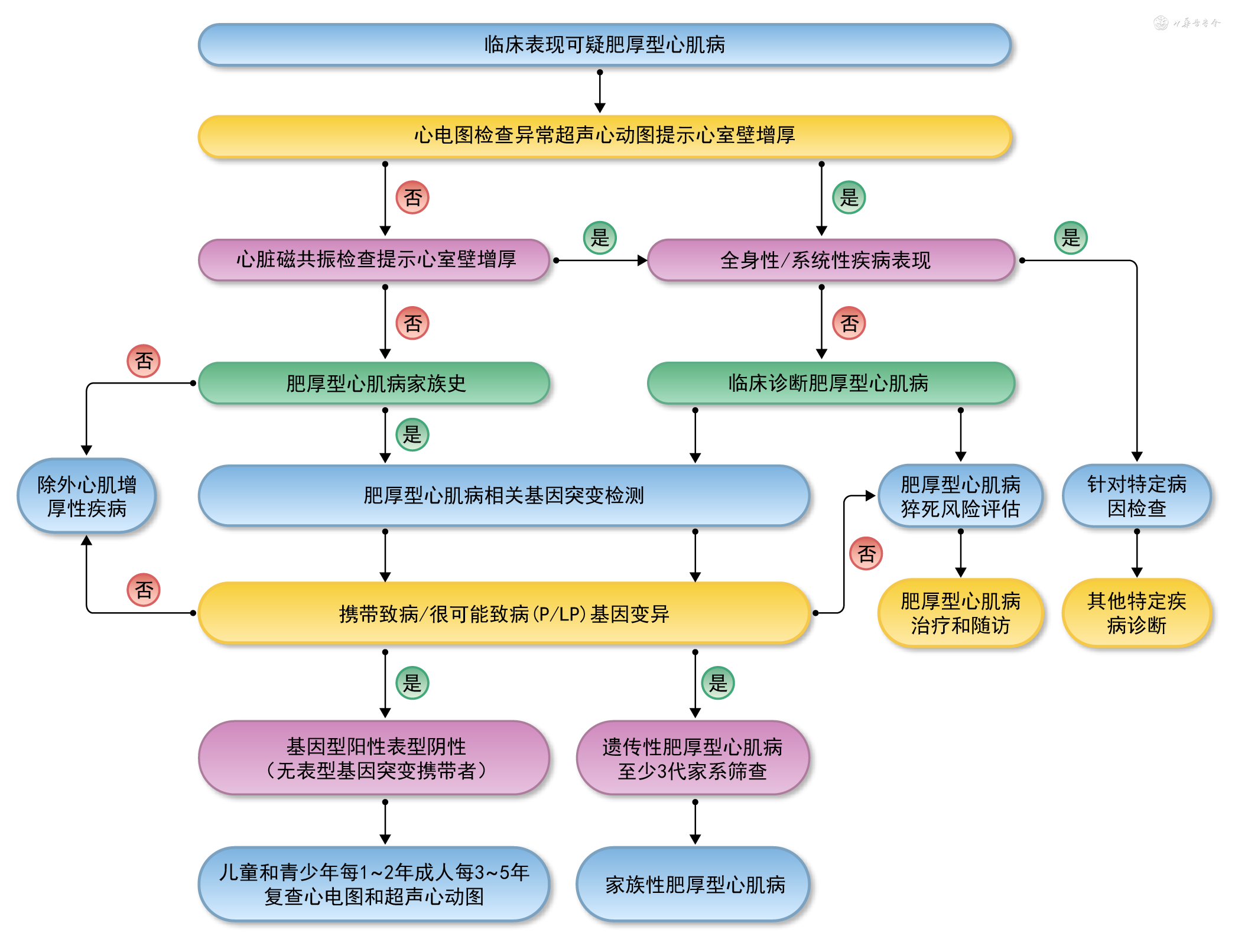
遗传评估通常包括遗传咨询和基因检测两部分内容。遗传评估首先针对家族中第一个被确诊为HCM的患者(称为“先证者”或“指征病例”)。
1. 遗传咨询:对于先证者,无论是否进一步开展相关的临床诊疗或基因检测,均推荐进行遗传咨询(Ⅰ类推荐,B级证据),称为检测前遗传咨询[1, 2, 3, 4]。主要达到两方面目的:(1)完善家族史调查及家系图谱绘制,为下一步的病因学检测提供证据和线索;(2)向准备行基因检测的患者及其亲属详细解释,使其充分认知进行基因检测发现遗传性心脏疾病的益处及疾病的潜在危害等。先证者进行基因检测后也要进行遗传咨询(Ⅰ类推荐,B级证据),称为检测后遗传咨询。也有两方面目的:(1)结合患者临床表型对基因检测结果进行专业解释,评估基因检测结果对于患者个人和家族成员危险分层的价值;(2)告知患者及亲属,需要根据基因变异致病性分类的进展情况,定期对基因检测结果进行再解释[85]。
2. 基因检测:详见“五、辅助检查,(四)基因检测”部分。
家系筛查应包括临床评估和基因检测两部分内容。临床评估内容包括症状评估和辅助检查,症状评估主要指活动相关的呼吸困难、胸痛、心悸或晕厥等,辅助检查至少包括12导联心电图及超声心动图检查。
目前推荐,对来自HCM基因型阳性家族的一级儿童亲属,推荐在其他家族成员诊断HCM时即应启动临床评估(包括心电图和超声心动图)和基因检测,以后每1~2年定期复查心电图和超声心动图;对于其他类型一级儿童亲属,推荐在家庭成员诊断HCM后的任何时候(但不晚于青春期)启动临床评估,以后每隔2~3年定期复查心电图和超声心动图[3],推荐临床评估至少持续到中年期(50岁)[3]。
根据先证者是否进行基因检测以及检测结果,开展相应的家系筛查策略如下[86, 87]。
1. 先证者检测到P/LP类型基因变异的家系:推荐开展级联性/预测性家系筛查,首先调查至少包括3代亲属的家族史,然后针对一级亲属启动临床评估和基因检测。(1)如果一级亲属携带与先证者相同的致病基因变异且具有HCM相关的临床表现,明确诊断HCM,进行规范治疗和常规随访。(2)如果一级亲属携带与先证者相同的致病基因变异但不具有LVH的证据且没有症状,诊断为“基因型阳性表型阴性”个体,推荐长期定期随访,监测疾病的进展。(3)如果亲属未检测到致病基因变异,初次临床评估也无HCM相关的临床表现,则可以解除进一步的临床监测。除非该家系出现新的临床状况,或基因变异类型再分类后降级为VUS或B/LP类型,则转为定期临床评估(图2)。
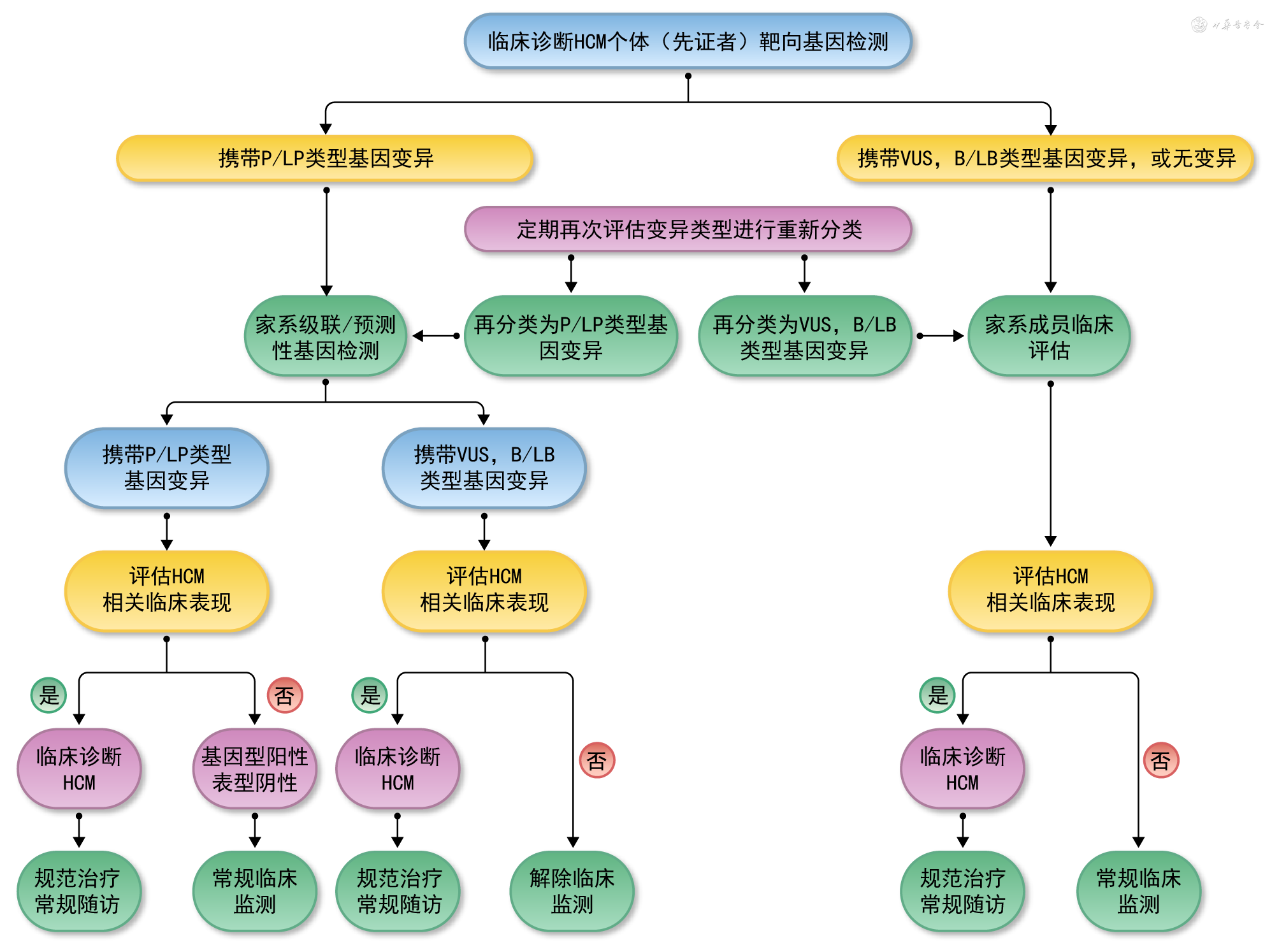
(注:HCM:肥厚型心肌病;P/LP:致病/很可能致病;VUS:意义未名的变异;B/LB:良性/很可能良性)
2. 先证者未检测到P/LP类型基因变异的家系:如果先证者尚未进行基因检测,或检测结果为阴性,或检测结果为意义未明的变异(variant of uncertain significance,VUS)或B/LB类型变异,建议亲属进行详细的临床评估。(1)如果发现HCM相关的临床表现,则诊断为HCM,进行规范治疗和常规随访。(2)如果初次评估未发现HCM相关的临床表现,则定期临床评估(图2)。
1. 定义:基因型阳性表型阴性(genotype positive phenotype negative,G+P-)个体,是指携带与HCM相关的P/LP类型基因变异(基因型阳性,G+),但是心脏影像学检查不存在心室壁增厚的证据(表型阴性,P-)且临床无症状的个体[10]。
2. 亚临床结构和(或)功能异常:尽管心脏影像学检查无LVH表现,但是部分(G+P-)个体可以有心电图异常,可以存在轻微的超声心动图异常,如心肌应变的改变、左心室舒张功能异常、二尖瓣叶拉长、CMR检查LGE阳性(心肌纤维化)、肌小梁异常、心肌隐窝以及左心房轻度扩大等[88, 89]。目前,这些亚临床结构和(或)功能改变的临床意义尚不明确。
3. 表型转化:研究发现,部分(G+P-)个体在随访过程中出现心室壁厚度增加,符合HCM的诊断标准(≥13 mm),出现了表型转化[90],发生率从6%~30%不等,从基因型阳性到表型阳性时间也不同。
4. 预后:(G+P-)个体只要没有发生表型转化,预后较好,多数无临床症状,发生SCD罕见。但是,部分个体可以发生表型转化,出现临床症状,甚至发生SCD。
心室壁增厚是HCM的典型特征,但有多种生理和病理因素可以导致心室壁增厚,称为HCM的“拟表型”,因此,在临床诊断HCM前需与其他疾病鉴别。
长期规律锻炼可以使心脏发生适应性改变,表现为左心室轻度对称性肥厚(通常≤15 mm),但是左心室舒张功能正常,心肺运动功能良好,无心肌病家族史,基因检测阴性。停止锻炼3个月后心肌肥厚程度可以减轻或消退。
一般高血压病史较长,长期血压控制不达标,通常为对称性轻度肥厚(≤15 mm),肥厚心肌呈均匀低回声,失代偿期左心室腔可增大。心电图可见左心室高电压,基因检测一般阴性。严格血压控制6~12个月后左心室心肌肥厚可以减轻或消退。
AS可以引起心脏后负荷增加,导致心肌代偿性肥厚,一般是轻度对称性肥厚(≤15 mm),与HCM存在以下区别:①收缩期杂音位置较高,以胸骨右缘第2肋间和胸骨左缘第3肋间明显,杂音向颈部传导,改变心脏前后负荷措施对杂音强度影响不大;②超声心动图检查可见主动脉瓣叶增厚、收缩期开放受限,瓣口面积缩小;③心导管检查提示左心室与主动脉之间存在收缩期压差,而左心室腔与LVOT之间无压差。
心脏淀粉样变可主要分为轻链型(light-chain cardiac amyloidosis,AL-CA)和转甲状腺素蛋白型(transthyretin cardiac amyloidosis,ATTR-CA)。由于淀粉样物质在心肌细胞外基质沉积,导致心室壁假性肥厚。通常为对称性心室壁增厚,但心电图表现为低电压或正常电压,QRS电压与室壁厚度比值下降;超声心动图可见室间隔和室壁均匀增厚,颗粒状回声增强,房间隔和瓣膜也可以增厚;CMR表现为心内膜下弥漫性甚至透壁性(全层)强化;具有以上警示信号的患者应高度怀疑心脏淀粉样变,需进行血、尿游离轻链及免疫固定电泳等检查用于AL-CA的筛查和诊断,焦磷酸盐放射性核素骨扫描以及转甲状腺素蛋白基因检测辅助ATTR-CA的诊断和分型;受累器官和/或心内膜活检病理学检查结果为心脏淀粉样变诊断和分型的金标准。可有心脏外表现:AL-CA可有50%~70%患者出现肾脏受累,表现为大量蛋白尿,不伴血尿;周围神经受累表现为四肢对称性感觉及运动功能障碍、腕管综合征等,自主神经受累主要表现为体位性低血压、排尿困难、排汗异常等;累及消化系统者可出现消化不良综合征及腹泻等临床表现;还可出现巨舌、眶周紫癜等特征性软组织受累表现;此外还可出现肝脏、皮肤、眼部受累等相关症状[91]。
1. Fabry病[92]:由GLA基因突变引起,遗传模式为X连锁隐性遗传,病因为α-半乳糖苷酶A的基因突变,导致其编码的α-半乳糖苷酶A功能部分或全部缺失,三聚己糖神经酰胺的正常降解受阻,未降解的底物在多种组织的细胞溶酶体中堆积,造成相关组织的功能障碍。心脏受累表现为心肌向心性肥厚,二尖瓣主动脉瓣增厚伴轻中度反流,乳头肌增厚,心电图常表现为左心室高电压及传导系统受累,也可见短PR间期不伴有WPW。CMR检查可见后侧壁中层LGE。其他系统受累表现如:血管角质瘤、肾脏损害、肢端感觉异常、少汗或无汗、早发脑梗、眩晕或听力障碍、角膜环形增生及胃肠症状等。α-半乳糖苷酶A酶活性测定及基因检测有助于明确诊断。
2. 糖原贮积症:常合并多器官受累的临床表现,主要包括以下两类:(1)Danon病[93]:是一种X连锁显性遗传病,由位于Xq24的LAMP2编码基因突变所致。起病早,男性常于20岁前,女性多于成年期发病。具有典型的三联征表型:心肌肥厚伴遗传性WPW、肌无力和智力发育迟缓。典型病理特征为骨骼肌细胞胞浆内空泡,免疫组化染色提示LAMP-2缺乏。可根据基因检测及特征性病理改变予以鉴别。(2)Pompe病[94]:婴儿型常累及心肌,多于出生后数月内发病,以心肌肥厚及重度全身性肌张力过低为主要特征,常于1岁前死亡。迟发型者累及心肌者罕见,以骨骼肌病为主,常表现为进行性肌无力。肌肉活检电镜下显示为空泡肌病伴溶酶体内糖原累积,且细胞质中有游离糖原。空泡过碘酸-希夫反应阳性,可被淀粉酶消化,且酸性磷酸酶染色阳性。
3. PRKAG2心脏综合征[95]:由编码单磷酸腺苷激活蛋白激酶γ2亚单位的PRKAG2基因突变引起,遗传模式为常染色体显性遗传。心脏受累表现为心肌肥厚、遗传性WPW、传导系统障碍及室上性心律失常四联征,其他系统表现较少或无。基因检测有助于明确诊断。
Friedreich共济失调[96]:是编码可溶性线粒体蛋白frataxin的FXN基因突变导致,遗传模式为常染色体隐性遗传,心脏受累主要表现为心肌肥厚。基因检测有助于明确诊断。
包括Noonan综合征[97]、LEOPARD综合征[98]等,统称为RAS病[99],分别由编码蛋白酪氨酸磷酸酶非受体11型的PTPN11基因及原癌基因c-Raf或RAF1及BRAF基因等突变引起,遗传模式为常染色体显性遗传。基因检测有助于诊断。
原发线粒体疾病是由核DNA或线粒体DNA突变所致,常见编码呼吸链蛋白复合物的基因突变,导致能量代谢障碍,出现多系统受累的症状,以对有氧代谢需求高的脑、骨骼肌及心肌表现为主。心脏受累表现以心肌肥厚最常见;其他系统受累表现包括神经肌肉病变、内分泌、消化系统或肾脏等。基因检测有助于确诊[100]。
HCM的治疗目标包括缓解临床症状,改善心脏功能,延缓疾病进展,减少疾病死亡。
1. 对于有症状的梗阻性HCM患者,使用药物治疗或侵入式治疗方式改善症状。
2. 对于有症状的非梗阻性HCM患者,主要针对合并症进行治疗。
3. 无症状HCM患者需要定期进行临床评估。即使存在LVOTO,也不推荐进行SRT。
4. 所有HCM患者都应常规开展SCD的风险评估和危险分层,进行相应预防和治疗。
药物治疗的主要目的是缓解HCM患者的症状,目前尚没有证据显示药物治疗可以改变HCM的自然病史。药物治疗的靶点是LVOTO,但LVOTO随日常活动变化明显,因此,评价药物治疗有效性主要是依据患者的症状反应,而不是根据测量LVOT压差的变化。
目前,症状性梗阻性HCM患者的药物治疗种类主要包括以下几种[1, 2, 3, 4]:
1. β受体阻滞剂:是最早被研究用于治疗HCM患者的药物,可以抑制心肌收缩力,降低LVOT压差(主要降低运动时LVOT压差),减轻LVOTO;可以减慢心率,改善心室舒张期充盈,明显改善患者的心功能和生活质量[101, 102, 103]。目前,β受体阻滞剂多作为一线治疗药物[1, 2, 3, 4]。对于症状性梗阻性HCM患者,推荐使用无血管扩张作用的β受体阻滞剂,包括普萘洛尔、美托洛尔和比索洛尔等,从小剂量起始,逐渐滴定至治疗有效(症状缓解)或最大耐受剂量(通常指静息心率达到55~60 次/min)(Ⅰ类推荐,B级证据)。儿童HCM患者也可以使用β受体阻滞剂。
2. 心肌肌球蛋白抑制剂:Mavacamten是选择性心肌肌球蛋白变构抑制剂,通过选择性降低心肌肌球蛋白重链的ATP酶活性,可逆地抑制肌球蛋白-肌动蛋白横桥的过量形成,同时促使整个肌球蛋白群体转向节能的超松弛状态,从而抑制心肌过度收缩、改善舒张顺应性及能量代谢[104]。EXPLORER-HCM研究[105]结果表明,Mavacamten 治疗组在复合功能主要终点方面的缓解率约为安慰剂组的两倍,同时显著降低运动后LVOT压差,也快速且持续降低静息和Valsalva激发LVOT压差。Mavacamten治疗组其他次要终点包括运动能力、心功能、症状和健康状况均较安慰剂组得到显著改善,心脏生物标志物N末端B型利钠肽原(N terminal-pro B type natriuretic peptide,NT-proBNP)和心肌肌钙蛋白I(cardiac troponin I,cTnI)水平也显著降低[106, 107]。VALOR-HCM研究[108]表明Mavacamten治疗16周明显减少了符合指南SRT指征的患者比例,给药物难治性严重梗阻性HCM患者提供了SRT治疗以外的新选择。非梗阻性HCM患者中进行的MAVERICK-HCM研究表明Mavacamten治疗可降低与室壁应力和心肌损伤相关的标志物(NT-proBNP和cTnI)水平。2022年4月获得美国食品和药物管理局(Food and Drug Administration,FDA)批准,是首个且唯一获批的心肌肌球蛋白抑制剂,可用于治疗症状性NYHA心功能Ⅱ~Ⅲ级的梗阻性HCM成人患者,以改善心功能和症状(Ⅰ类推荐,B级证据)。
Aficamten(CK-274/CK-3773274)是一种新型选择性小分子心肌肌球蛋白抑制剂,其药动学特性支持每日1次的给药计划。REDWOOD-HCM结果[109]表明,aficamten相比安慰剂可显著降低LVOTG和NT-proBNP水平。目前,FDA和中国国家药品监督管理局(National Medical Products Administration,NMPA)-药品审评中心(Center for Drug Evaluation,CDE)均已授予aficamten治疗症状性梗阻性HCM的“突破性治疗药物”认定。
对于LVEF<55%的患者不建议使用心肌肌球蛋白抑制剂。如果用药过程中出现心衰恶化表现或LVEF<50%,建议暂停使用。
3. 非二氢吡啶类钙离子通道阻断剂(calcium channel blocker,CCB):具有负性肌力和负性频率作用,可以减轻LVOTO,改善舒张期心室充盈,改善患者症状。对于β受体阻滞剂治疗无效、无法耐受或有禁忌的症状性梗阻性HCM患者,推荐使用非二氢吡啶类CCB[1, 2, 3, 4],包括维拉帕米(Ⅰ类推荐,B级证据)或地尔硫卓(Ⅰ类推荐,C级证据)。儿童和青少年HCM患者也可以使用维拉帕米。但是,对于静息时存在严重呼吸困难或心衰体征,低血压或心原性休克,病态窦房结综合征,或二度或三度房室传导阻滞(除非已植入心脏起搏器),静息LVOT压差明显升高(>80~100 mmHg)的患者,不推荐使用非二氢吡啶类CCB(Ⅲ类推荐,C级证据)。
4. 丙吡胺:属于Ⅰa类抗心律失常药物,同时具有较强的负性肌力作用,可以抑制心肌收缩力,减轻SAM现象和MR程度,可以降低LVOT压差[110, 111]。对于使用β受体阻滞剂和非二氢吡啶类CCB后仍有与LVOTO相关的持续严重症状的患者,推荐加用丙吡胺,与β受体阻滞剂或非二氢吡啶类CCB联合应用并逐渐滴定至最大耐受剂量(Ⅰ类推荐,B级证据)。
5. 西苯唑啉:也属于Ⅰa类抗心律失常药物,可以明显降低LVOT压差,减轻LVOTO;还可以改善左心室舒张功能[112]。对于使用β受体阻滞剂和非二氢吡啶类CCB后仍然有症状的患者,推荐加用西苯唑啉,与β受体阻滞剂或非二氢吡啶类CCB联合应用,并逐渐滴定至最大耐受剂量(Ⅰ类推荐,B级证据)。
6. 其他[1, 2, 3, 4]:(1)伴有严重激发性LVOTO表现为急性低血压和肺水肿,是一种急危重症,必须及时识别,治疗上如果对补充液体治疗无反应,推荐静脉内使用苯肾上腺素或其他无正性肌力作用的血管收缩剂(如去甲肾上腺素),或者联合口服或静脉β受体阻滞剂(Ⅰ类推荐,C级证据)。(2)梗阻性HCM患者经其他药物治疗后如果持续存在呼吸困难症状,同时存在容量超负荷或者心腔内压力增高的临床表现,可以考虑谨慎使用小剂量口服袢利尿剂或噻嗪类利尿剂,以减轻患者的劳力性呼吸困难症状(Ⅱb类推荐,C级证据)。(3)对于症状性梗阻性HCM患者,不推荐使用正性肌力药物(如洋地黄类、磷酸二酯酶抑制剂、β1受体激动剂等)、动脉及静脉血管扩张剂(如血管紧张素转化酶抑制剂、血管紧张素受体阻断剂、二氢吡啶类CCB、硝酸酯类药物)、大剂量利尿剂(Ⅲ类推荐,C级证据)。
侵入式治疗措施包括外科室间隔心肌切除术(SSM)、室间隔心肌消融术(septal myocardial ablation,SMA)和双腔起搏器植入术,前两种治疗方式可以使室间隔变薄,故统称为SRT。
1. SRT的适应证:(1)临床标准:患者经充分药物治疗后仍然存在严重呼吸困难或胸痛症状(NYHA心功能Ⅲ级或Ⅳ级),或者存在其他与活动相关的症状(如反复发生晕厥或晕厥前状态),与LVOTO相关,影响患者日常活动和生活质量。(2)血流动力学标准:静息或激发时LVOT峰值压差≥50 mmHg,与室间隔肥厚或二尖瓣SAM现象相关。(3)解剖标准:由术者个人判断拟行手术的室间隔厚度是否能足够安全有效地进行操作。对于无症状且活动耐力正常的梗阻性HCM患者或者虽然有症状但经优化药物治疗可以控制,则不推荐进行SRT(Ⅲ类推荐,C级证据)。
2. SSM:(1)手术指征及推荐意见[1, 2, 3, 4]:①符合SRT适应证,能够接受心脏外科手术的患者,推荐进行SSM(Ⅰ类推荐,B级证据)[113, 114, 115, 116, 117];②合并需要外科手术治疗的相关心脏疾病(如相关的乳头肌异常、二尖瓣前叶显著拉长、二尖瓣原发病变、多支冠状动脉病变、主动脉瓣狭窄等)的患者,也推荐进行SSM(Ⅰ类推荐,B级证据)[116,118];③SMA治疗失败(残余梗阻)或术后复发的患者,推荐进行SSM(Ⅰ类推荐,C级证据)[119, 120]。④符合SRT适应证,经过详细评估后提示存在并非单独由二尖瓣SAM现象引起的中度至重度MR,应该考虑行二尖瓣修复术(首选)或置换术(Ⅱa类,C级)。不推荐仅仅以缓解LVOTO为目的进行二尖瓣置换术(Ⅲ类推荐,B级证据)。(2)主要术式及发展历史:开胸、经主动脉切口室间隔心肌切除术,是治疗梗阻性HCM的标准手术方法,最早由Morrow[7]等人于1961年报道。目前,Morrow手术扩大了室间隔切除范围,远端超越二尖瓣叶室间隔接触部位,达到乳头肌基底部甚至心尖部,称为扩大的室间隔心肌切除术或改良的Morrow手术[121, 122]。目前,除了室间隔肥厚引起LVOTO主要经主动脉切口行室间隔心肌切除术外,其他特殊类型梗阻性HCM,如ApHCM,可以经心尖切口行心肌切除术[123];对于MVOHCM,可以经心尖切口或(或)主动脉切口行心肌切除术[124]。(3)治疗效果:SSM可以快速明显减轻LVOTO,使MR减轻或消失。可以使患者的远期生存率接近正常人。国外研究显示,症状性梗阻性HCM患者SSM术后1年、5年及10年生存率分别为98%、96%及83%,与普通人群之间差异无统计学意义,但明显优于未接受SSM的患者(1年、5年及10年生存率分别为90%、79%及61%)[113]。我国研究结果显示,症状性梗阻性HCM患者SSM术后1年及5年累积生存率分别为99%和97%[116]。(4)安全性及并发症:目前,单纯SSM的手术相关病死率<1%[115],而SSM联合二尖瓣手术的病死率为3%~5%。其他术后早期常见并发症包括完全性房室传导阻滞(atrioventricular block,AVB)、左束支传导阻滞(left bundle branch block,LBBB)、房颤、室间隔穿孔及主动脉瓣关闭不全等。
3. 经皮腔内室间隔心肌消融术(percutaneous transluminal septal myocardial ablation,PTSMA):(1)适应证及推荐意见:①符合SRT的适应证,但是由于高龄或严重合并症,考虑外科手术高危或禁忌而不能接受外科手术,或者不愿接受外科手术的患者,推荐进行PTSMA(Ⅰ类推荐,C级证据)[1, 2, 3, 4,125],同时要求梗阻位于室间隔基底段,冠状动脉造影检查提示间隔支动脉适合行PTSMA。②对于SSM治疗失败(残余梗阻)或PTSMA术后复发的患者,可以考虑再次进行PTSMA(Ⅱb类推荐,C级证据)[126]。(3)治疗方式:目前主要采用ASA,这是最经典的PTSMA,利用无水酒精(96%~99%乙醇)化学消融、阻断间隔支动脉,造成区域心肌坏死,从而消除室间隔肥厚,降低LVOTO。(4)治疗效果:PTSMA可以显著降低LVOT峰值压差,减轻LVOTO,减轻或消除MR,从而改善患者症状和心功能,提高活动耐力和生活质量。通常将LVOT峰值压差下降≥50%或LVOT峰值压差<30 mmHg作为PTSMA操作成功的标志。PTSMA也可以显提高症状性梗阻性HCM患者的远期生存率,使其接近年龄、性别匹配的普通人群[127, 128]。(5)安全性及并发症:ASA术后主要不良心脏事件发生率<2%。围术期并发症主要包括:AVB(需要植入心脏永久起搏器的比例<10%),右束支传导阻滞(right bundle branch block,RBBB)(第一间隔支动脉供血),心肌梗死及心肌瘢痕诱导的室性心律失常等。
4. SSM与ASA比较:目前,SSM与ASA均是治疗梗阻性HCM的有效方式,在减轻LVOTO、改善症状和生活质量、短期及长期病死率方面无显著差别[129]。只是ASA术后残余梗阻的发生率更高,可能需要再干预的概率更高。一项对3 859例梗阻性HCM患者的分析结果显示,在对年龄、性别及合并症进行调整后,与SSM比较,ASA的10年全因死亡风险增加68%[130]。在临床实践中,需要根据患者年龄、梗阻部位及程度、伴随疾病及患者意愿等方面因素综合评价后做出个体化选择。(1)年龄方面,成人患者只要能够耐受心脏外科手术,一般首选SSM,除非高龄、因各种原因不能耐受外科治疗,ASA作为替代治疗方式。(2)梗阻程度方面,ASA主要适用于室间隔基底部轻中度梗阻(室壁最大厚度<30 mm)的患者;对于左心室极度肥厚(室壁最大厚度≥30 mm)或者严重LVOTO(静息LVOT峰值压差≥100 mmHg)患者,一般首选SSM。(3)伴随疾病:①对合并其他需外科治疗疾病患者,首选SSM。如果合并冠状动脉单支病变(前降支),可先行ASA再行冠状动脉介入治疗。②外科术后LBBB发生率高,若患者术前已存在RBBB,术后易出现三度AVB,需要植入永久心脏起搏器,因此外科治疗要慎重;相反,ASA术后RBBB发生率高,若患者术前已存在LBBB,则需慎重选择ASA。
5. 其他SMA:(1)经皮心肌内室间隔射频消融术(percutaneous intramyocardial septal radiofrequency ablation,PIMSRA,又称Liwen术式)[131, 132]:可以考虑在有治疗经验的中心作为SSM或ASA的一种替代治疗方式,其远期疗效值得进一步研究。(2)心内膜室间隔射频消融术(endocardial radiofrequency ablation of septal hypertrophy,ERASH)[133, 134]:ERASH并发症发生率较高,远期疗效需要进一步观察。
6. 双腔起搏器植入术:目前双腔起搏器(右心房-右心室顺序起搏)治疗梗阻性HCM患者中的应用较少,远期疗效不确切,仅对于SSM或ASA术后发生心脏传导阻滞风险高危的患者,可以考虑植入双腔起搏器(Ⅱb类推荐,C级证据)[4]。
HCM患者常有胸闷、气短等呼吸困难症状,主要是由左心室舒张功能异常引起,表现为HFpEF;少数进展至ES-HCM(LVEF<50%),表现为HFrEF或HFmrEF。
药物治疗的目标是控制心率以降低左心室舒张末压,改善左心室充盈。推荐使用β受体阻滞剂和非二氢吡啶类CCB作为一线治疗药物(Ⅰ类推荐,C级证据)。使用上述药物后仍有劳力性呼吸困难或者有液体潴留表现的患者,应该考虑谨慎使用小剂量利尿剂(袢利尿剂或噻嗪类利尿剂)(Ⅱa类推荐,C级证据)。
2. ES-HCM的治疗:(1)药物治疗:参考最新心衰指南意见[135, 136],推荐给予指南指导的药物治疗(guideline directed medical therapy,GDMT)(Ⅰ类推荐,C级证据),应该考虑停用负性肌力作用药物,如非二氢吡啶类CCB、丙吡胺、西苯唑啉等(Ⅱa类推荐,C级证据)。(2)植入式心脏转复除颤起搏器(ICD):ES-HCM患者SCD风险较高,可以考虑植入ICD(Ⅱa类推荐,C级证据)。(3)心脏再同步化治疗(cardiac resynchronization therapy,CRT):ES-HCM患者如果合并LBBB且QRS间期>130 ms,窦性心律,给予GDMT后仍有症状(NYHA心功能Ⅱ~Ⅳ级),可以考虑应用CRT改善症状(Ⅱa类推荐,C级证据)。
3. 终末期心衰的治疗:(1)心脏移植:GDMT后仍有严重症状(NYHA心功能分级Ⅲ~Ⅳ级)或者反复发作致命性室性心律失常的HCM患者(不论LVEF水平),推荐根据最新的心脏移植等待标准[137]进行心脏移植评估(Ⅰ类推荐,B级证据),包括进行CPET评估患者心功能(Ⅰ类推荐,B级证据)。HCM患者心脏移植后的长期预后与非HCM的患者类似。(2)心室辅助装置(ventricular assist device,VAD):对于部分合并终末期心衰的HCM患者,在进行心脏移植前应该考虑进行植入VAD作为过渡(Ⅱa类推荐,B级证据),以减少等待心脏移植期间的死亡。
1. HCM合并房颤(房扑)的治疗:(1)抗凝治疗:房颤(房扑)显著增加HCM患者卒中和周围栓塞风险[78],与CHA2DS2-VASc评分结果无关[138]。因此,建议所有合并房颤(房扑)的HCM患者,只要没有禁忌证,均应给予抗凝治疗,不用依据CHA2DS2-VASc评分结果(Ⅰ类推荐,B级证据)。推荐将直接作用口服抗凝药(direct-acting oral anticoagulants,DOACs)作为一线治疗,口服维生素K拮抗剂(华法林)作为二线治疗(Ⅰ类推荐,B级证据)[139]。(2)心率控制策略:对于选择心率控制策略的HCM合并房颤患者,推荐根据患者的倾向和合并症情况使用β受体阻滞剂,或非二氢吡啶类CCB(Ⅰ类推荐,C级证据)。若药物治疗难以控制心室率,或出现不能耐受的药物不良反应,可以考虑行房室结消融术来控制心室率,改善症状(Ⅱb类推荐,C级证据)[4]。(3)节律控制策略:主要措施包括抗心律失常药物(antiarrhythmic drugs,AADs)、直流电复律、导管消融术及外科迷宫手术等。HCM合并房颤(房扑)应该考虑应用AADs(如胺碘酮)转复窦性心律(Ⅱa类推荐,B级证据)[3, 4];应该考虑应用胺碘酮或其他AADs(如丙吡胺、索他洛尔等)维持窦性心律(Ⅱa类推荐,C级证据)[4]。伴有血流动力学不稳定或严重的心绞痛或心衰症状,应该考虑直流电复律恢复窦性心律(Ⅱa类推荐,C级证据)[2, 3, 4]。药物治疗难以控制症状的房颤,或对药物治疗不耐受(禁忌)或不接受的HCM患者,应该考虑导管消融术(Ⅱa类推荐,B级证据)[140]。外科迷宫手术是治疗房颤的一种有效措施,尤其是进行SSM的HCM患者,应该考虑同期进行外科迷宫术(Ⅱa类推荐,B级证据)[141]。
2. HCM合并室性心律失常的治疗:(1)药物治疗:推荐首选β受体阻滞剂治疗(Ⅰ类推荐,C级证据)[1, 2, 3, 4]。使用β受体阻滞剂后如果仍存在症状性室性心律失常,或者植入ICD后反复放电,推荐联合使用AADs,包括胺碘酮(Ⅰ类推荐,B级证据),或索他洛尔、多非利特、美西律等(Ⅰ类推荐,C级证据)[142]。(2)非药物治疗:推荐植入ICD治疗,可以预防致命性的室性心律失常导致的SCD。植入ICD后需优化药物治疗和起搏器程控,减少恰当或不恰当的放电风险(Ⅰ类推荐,B级证据)。如果反复发作局灶起源的症状性、持续性、单形性VT,应该考虑导管消融治疗(Ⅱa类推荐,C级证据)。(3)心脏移植:反复发作致命性室性心律失常的HCM患者(不论LVEF水平),推荐根据最新的心脏移植等待标准进行心脏移植评估(Ⅰ类推荐,B级证据)。
HCM患者的治疗流程图见图3。
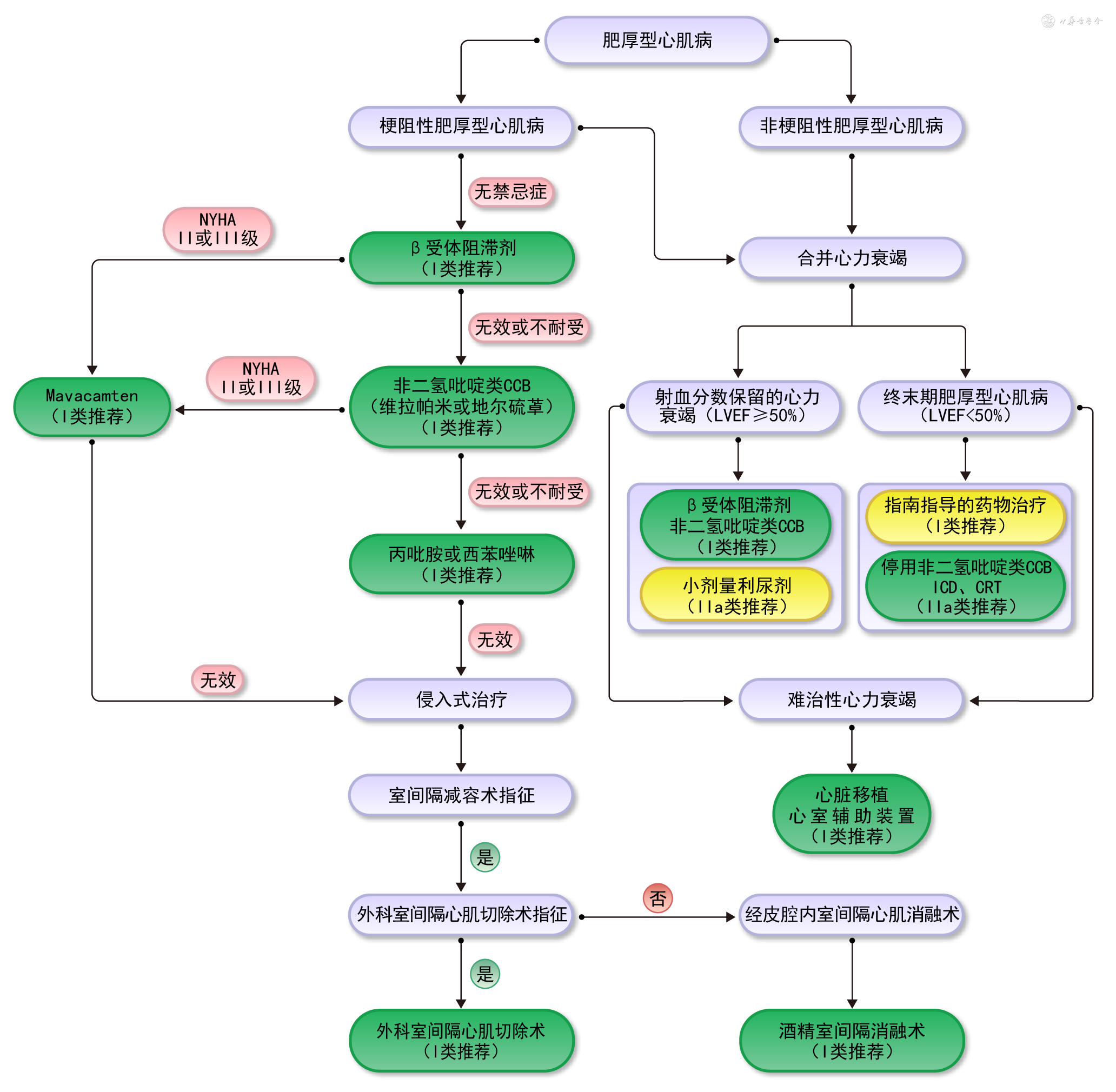
(注:HYHA:纽约心脏协会;CCB:钙离子通道阻断剂;LVEF:左心室射血分数;ICD:植入式心脏转复除颤起搏器;CRT:心脏再同步化治疗)
研究显示,SCD是HCM患者的首位死亡原因,也是HCM最严重的并发症之一,最常见的病理生理机制是致命性室性心律失常。因此,建议HCM患者在初诊时即应进行SCD风险评估,以后每隔1~2年或临床状况发生变化时再次评估,根据SCD的风险高低,决定预防和治疗措施。目前,ICD是预防和治疗SCD的最有效措施。
目前研究报道的成人HCM患者SCD的危险因素至少包括以下几种[1, 2, 3, 4]:(1)既往心脏骤停或持续性(持续时间>30s)室性心律失常导致的血流动力学不稳定病史;(2)早发HCM相关SCD家族史,即≥1个一级亲属在年龄≤50岁发生SCD、心脏骤停或持续性室性心律失常,明确或很可能由HCM引起;(3)近期(6个月内)发生不明原因晕厥,考虑可能由心律失常引起(非神经介导性的血管迷走性晕厥);(4)极度左心室肥厚(左心室最大室壁厚≥30 mm);(5)LVAA;(6)ES-HCM(LVEF<50%);(7)24~48h动态心电图检查发现NSVT;(8)CMR检查提示存在广泛心肌纤维化(≥15%左心室质量)。
1. 目前,临床应用最广泛的关于成人HCM患者5年SCD风险的预测模型是HCM Risk-SCD模型[143],多部指南或共识推荐该模型用于HCM患者SCD的风险预测。根据该预测模型,如果5年SCD风险≥6%,定义为高危组;5年SCD风险≥4%但<6%,定义为中危组;5年SCD风险<4%,定义为低危组。但是,该模型仅适用于年龄≥16岁,既往没有心脏骤停或持续性室性心律失常病史的成年HCM患者,不适用于年龄<16岁的HCM患儿;对于最大心室壁厚度≥35 mm的HCM患者也存在低估风险的可能;模型也没有纳入新的SCD危险因素,如ES-HCM、左心室心尖部室壁瘤及CMR-LGE等。
2. 强化的美国心脏病学会(American College of Cardiology,ACC)和美国心脏协会(American Heart Association,AHA)策略,2019年发布了纳入关于成人HCM患者发生SCD风险的7个主要危险因素,包括早发HCM相关家族史、近期不明原因晕厥、左心室肥厚(尤其左心室最大室壁厚度≥30 mm)、NSVT、LVEF<50%、LVAA以及CMR-LGE检测的心肌纤维化[144]。根据是否存在上述危险因素,将研究对象分为高危人群和低危人群。
1. 危险因素:目前,文献报道的与儿童(年龄<18岁)HCM患者SCD相关的主要危险因素包括以下4点[1, 2, 3, 4]:(1)早发的HCM相关的SCD家族史;(2)不明原因晕厥的个人史;(3)极度左心室肥厚(最大室壁厚度≥30 mm或z值>6);(4)动态心电图检查发现NSVT。目前,LVAA、ES-HCM以及CMR检查提示存在广泛心肌纤维化等危险因素在儿童中的研究较少,预测价值不确定。
2. 预测模型:针对儿童HCM患者SCD的预测模型目前主要有2个,一个是由欧洲学者提出的HCM Risk-kids预测模型[145],纳入变量包括最大室壁厚度、左心房内径、NSVT及LVOT瞬时峰值压差等;另一个是由美国学者提出的PRIMACY预测模型[146],纳入变量包括诊断时年龄、室间隔厚度、LVPWT、左心房内径、LVOT最大瞬时峰值压差、NSVT及晕厥史。目前,这两个预测模型的预测价值都需要更多研究进行外部验证。
既往明确发生过心脏骤停或致命性室性心律失常导致血流动力学紊乱的HCM患者(包括成人和儿童患者),推荐植入ICD进行二级预防(Ⅰ类推荐,B级证据)。
成人HCM患者,存在上述5项(危险因素2~6)成人SCD危险因素至少一项或SCD风险预测模型评估为高危,应该考虑植入ICD进行一级预防(Ⅱa类推荐,B级证据)。如果无上述危险因素,但是动态心电图检查发现NSVT,或CMR检查提示存在广泛心肌纤维化,或SCD风险预测模型评估为中危,可以考虑植入ICD进行一级预防(Ⅱb类推荐,B级证据)。无上述危险因素的HCM患者,不推荐植入ICD(Ⅲ类推荐,B级证据)(图4)。成人HCM患者植入ICD前需要全面、系统评价SCD的风险高低,同时仔细评估植入ICD的潜在并发症,充分尊重患者的知情同意和决策权(Ⅰ类推荐,C级证据)[147]。
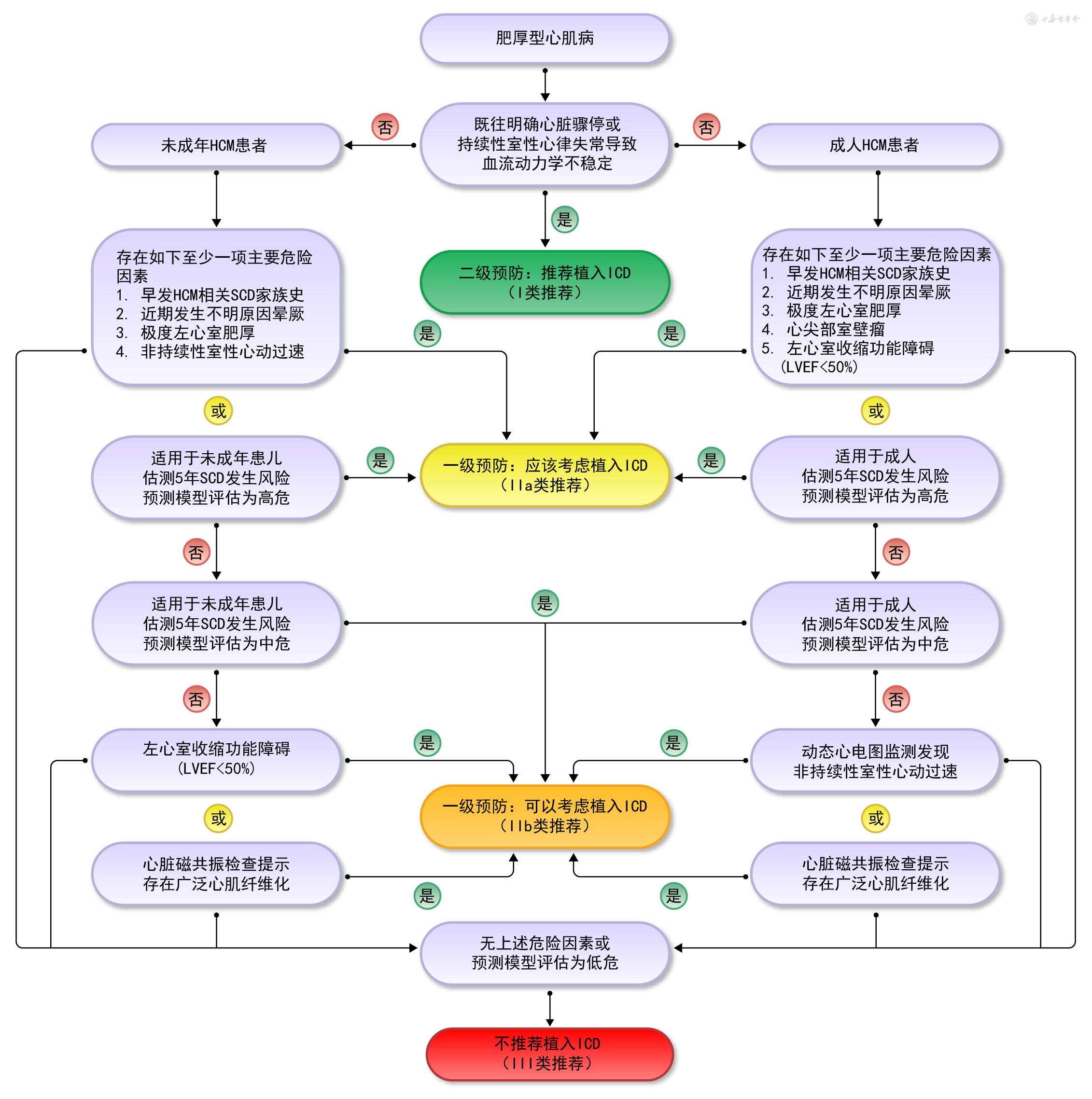
(注:HCM:肥厚型心肌病;ICD:植入式心脏转复除颤起搏器;LVEF:左心室射血分数;SCD:心脏性猝死)
儿童HCM患者,存在上述4项儿童SCD危险因素至少一项,或者SCD风险预测模型评估为高危,应该考虑植入ICD进行一级预防(Ⅱa类推荐,B级证据)。如果没有上述危险因素,但是LVEF<50%,或CMR检查提示存在广泛心肌纤维化,或SCD风险预测模型评估为中危,可以考虑植入ICD进行一级预防(Ⅱb类推荐,C级证据)。对于无上述危险因素的HCM患者,不推荐植入ICD(Ⅲ类推荐,B级证据)(图4)。儿童HCM患者在植入ICD前更需要全面、充分评估SCD风险及潜在并发症,与患儿及家长(或监护人)充分沟通、权衡利弊后决定(Ⅰ类推荐,C级证据)[147]。
1. 孕前和产前评估和咨询:妊娠会引起血浆容量和心输出量增加、全身血管阻力降低和高凝状态,使HCM孕妇及胎儿的风险增加,尤其是合并LVOTO和左心室舒张功能异常患者,对容量负荷的耐受性较差。因此,HCM女性患者在孕前和产前应进行充分的风险评估和咨询[148]。主要采用超声心动图检查评估心脏功能、MR及LVOTO情况。发生心脏事件的主要是孕前已经存在症状或既往发生过心脏事件的女性,因此,有症状的HCM女性应该在孕前采取积极措施以改善症状。
2. 孕期管理:(1)孕期监测和评估:HCM女性的孕期监测和评估重点包括症状、心脏功能、LVOTO以及心律失常等。(2)孕期药物治疗:孕期用药需要兼顾孕妇病情需要及药物对胎儿可能产生的影响。①β受体阻滞剂:多数β受体阻滞剂(包括美托洛尔、比索洛尔、拉贝洛尔、普萘洛尔)在孕期使用是安全的,但不建议使用阿替洛尔,有潜在胎儿风险。孕期使用β受体阻滞剂,需密切监测胎儿生长及胎儿心率。②非二氢吡啶类CCB和丙吡胺:孕期尽量避免使用。③AADs:多数AADs有潜在致畸作用,孕期禁忌使用。胺碘酮有导致胎儿甲状腺毒性、生长发育迟缓和心动过缓风险等,孕期应该尽量避免使用。④抗凝治疗:对于合并房颤或其他抗凝指征的HCM女性,可以在妊娠早期(前3个月)和36周以后使用低分子肝素抗凝,或者在妊娠中晚期使用低剂量(<5 mg/d)华法林抗凝治疗。目前关于DOACs在孕期使用的安全性数据有限,不建议使用。
HCM是一种高度异质性的心脏疾病,从婴儿到老年人均有可能发病。目前,成人HCM患者年病死率为0.5%~1.0%,主要死因包括SCD、心衰及房颤导致的卒中等[1, 2, 3, 4,10,150]。其中,年轻患者的死因主要是SCD,老年患者的死因主要是HCM相关的心衰和卒中。大多数HCM患者需要密切随访。
执笔专家:张健,张宇辉,邹长虹(中国医学科学院阜外医院)
指导专家:黄峻(江苏省人民医院),郑哲(中国医学科学院阜外医院)
专家组成员(按拼音首字母排序):艾力曼·马合木提(新疆医科大学第一附属医院),白玲(西安交通大学第一附属医院),陈还珍(山西医科大学第一医院),陈牧雷(首都医科大学附属北京朝阳医院),陈维(上海市第十人民医院),崔炜(河北医科大学第二医院),丁文惠(北京大学第一医院),董建增(首都医科大学附属北京安贞医院),董蔚(中国人民解放军总医院),范慧敏(同济大学附属东方医院),方理刚(北京协和医院),高传玉(阜外华中心血管病医院),格桑罗布(西藏自治区人民医院),郭延松(福建省立医院),韩秀敏(北部战区总医院),洪华山(福建医科大学附属协和医院),黄洁(中国医学科学院阜外医院),黄峻(江苏省人民医院),黄晓红(中国医学科学院阜外医院),季晓平(山东大学齐鲁医院),金玮(上海交通大学医学院附属瑞金医院),孔洪(四川省人民医院),黎励文(广东省人民医院),骆雷鸣(中国人民解放军总医院),李殿富(南京医科大学第一附属医院),李广平(天津医科大学第二医院),李应东(甘肃中医药大学附属医院),李勇(复旦大学附属华山医院),李悦(哈尔滨医科大学附属第一医院),李忠佑(北京大学人民医院),梁延春(北部战区总医院),刘斌(吉林大学第二医院),刘莹(大连医科大学附属第一医院),罗素新(重庆医科大学附属第一医院),马爱群(西安交通大学第一附属医院),马根山(东南大学附属中大医院),马晓昌(中国中医科学院附属西苑医院),毛静远(天津中医药大学第一附属医院),毛威(浙江省中医院),聂宇(中国医学科学院阜外医院),任景怡(中日友好医院),宋春莉(吉林大学第二医院),宋江平(中国医学科学院阜外医院),宋云虎(中国医学科学院阜外医院),宋昱(泰达国际心血管病医院),宋治远(陆军军医大学西南医院),沈涤非(武汉大学人民医院),孙艺红(中日友好医院),孙志军(中国医科大学附属盛京医院),唐其柱(武汉大学人民医院),陶蓉(上海交通大学附属瑞金医院),田庄(北京协和医院),童嘉毅(东南大学附属中大医院),佟倩(吉林大学第一医院),王江(陆军军医大学附属新桥医院),王涟(南京大学医学院附属鼓楼医院),王水云(中国医学科学院阜外医院),王祖禄(北部战区总医院),王宇石(吉林大学第一医院),王焱(厦门大学附属心血管病医院),徐大春(上海市第十人民医院),许顶立(南方医科大学南方医院),杨萍(吉林大学中日联谊医院),杨伟宪(中国医学科学院阜外医院),杨志明(山西医科大学第二医院),姚亚丽(兰州大学第一医院),余静(兰州大学第二医院),袁璟(华中科技大学同济医学院附属协和医院),张健(中国医学科学院阜外医院),张梅(山东大学齐鲁医院),张萍(北京清华长庚医院),张庆(四川大学华西医院),张瑶(哈尔滨医科大学附属第二医院),张宇辉(中国医学科学院阜外医院),赵兴胜(内蒙古自治区人民医院),赵燕(云南省第一人民医院),郑昭芬(湖南省人民医院),周蕾(江苏省人民医院),周胜华(中南大学湘雅二医院),周洲(中国医学科学院阜外医院),邹云增(复旦大学附属中山医院)
学术秘书:庄晓峰 田鹏超 李心晴 周琼 刘慧慧 周萍 翟玫 黄燕 王锦溪 韩慧侨
所有作者均声明不存在利益冲突
























