
炎症性肠病(IBD)是一类异质性的慢性弥漫性肠道炎症,疾病发生发展与免疫失调密切相关。树突状细胞(DC)在黏膜免疫中起到重要的作用,生理状态下可发挥免疫耐受功能,但在IBD中会抑制免疫耐受并触发过度的炎性免疫反应。然而DC的免疫失衡作用及其进一步启动的免疫信号通路尚不明确。RhoA作为免疫细胞分化和功能的关键调节剂,与DC的形态、增殖、抗原摄取、细胞迁移及抗原提呈等功能密切相关,RhoA信号通路异常可能影响DC的功能变化。本文针对DC及细胞内RhoA信号通路在IBD发病中的作用进行综述,希望对临床治疗有所启发和帮助。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
炎症性肠病(inflammatory bowel disease,IBD)是一种病因未明的胃肠道慢性炎症性疾病,其临床特征、组织病理学表现及免疫抑制剂的疗效都表明免疫因素在发病机制中起关键作用,针对调节免疫细胞的生物治疗和细胞内信号传导通路的靶向治疗也成为研究的热点。而树突状细胞(dendritic cell,DC)在黏膜免疫过程中起到至关重要的作用。DC作为最有效的抗原提呈细胞(antigen presenting cell,APC),可以摄取加工抗原并将其迁移至淋巴结,将抗原呈递给T、B细胞,诱导相应淋巴细胞分化,产生不同的免疫应答。在正常黏膜免疫中,DC诱导分泌抗原特异性IgA的B细胞和调节性T细胞(Treg)的产生,维持对自身抗原的免疫耐受,同时保持针对入侵病原体的主动免疫应答能力[1]。而在IBD中,DC针对入侵病原菌的免疫控制发生失调,导致以Th1、Th17等占主导地位的过度炎症反应[2,3],损伤肠壁,破坏肠黏膜屏障。RhoA作为免疫细胞分化和功能的关键调节剂,与DC的形态、增殖、抗原摄取、细胞迁移及抗原提呈等功能密切相关。DC功能变化可能与细胞中RhoA信号通路异常有关。过去的研究主要集中于适应性免疫在IBD发病中的作用,较少涉及DC的免疫功能。本综述将特别关注DC及其RhoA信号通路在IBD发病中的潜在作用,为研究和治疗这一疾病提供新的切入点。
在生理状态下,人体肠上皮细胞(intestinal epithelial cells,IECs)会在黏膜固有层分泌"镇静因子",包括视黄酸和转化生长因子-β(transforming growth factor-β,TGF-β),造成一种致耐受性的环境,因此DC表现为耐受或调节性表型[4],在抗原呈递时DC显示出主要组织相容性复合物(major histo-compatibility complex,MHC)Ⅱ类分子及共刺激分子的表达降低和(或)产生调节性细胞因子如白细胞介素(interleukin,IL)-10的能力增强,诱导非炎性淋巴细胞的发育[1]。此时,DC激活免疫调节机制并产生低水平免疫反应,从而控制炎症,例如产生具有调节特性的Treg和分泌IgA的B细胞,形成对共生菌群及食物抗原的整体耐受状态[1]。研究发现固有层DC中存在一类特殊亚群CD103+CX3CR1-DC,可激活CD4+ T细胞,促进TGF-β和视黄酸的分泌,诱导FOXP3+ Treg的产生,促使免疫耐受[5,6]。CD103+DC的迁移取决于其表面趋化因子受体CCR7的表达,虽然病原体或其他炎性刺激会增强其表达,但是这种表达受环境的影响较小。据估计,在无病原体感染的情况下,每天有5% ~ 10%的CD103+DC亚群迁移至肠系膜淋巴结[7]。可见在正常情况下,DC可使黏膜免疫系统长期处于免疫耐受状态。
在IBD患者的肠黏膜组织中,细胞间紧密连接的完整性受到影响,上皮屏障被破坏,病原体通过上皮层进入,并被位于IECs基底外侧膜上的模式识别受体(pattern recognition receptor,PRR)识别。在这种情况下,IECs会阻止"镇静因子"的分泌。此外,固有层中大量的促炎性细胞因子也会掩盖IECs分泌的镇静信号,DC将捕获的抗原识别为入侵的病原体,转变为促炎表型,从而阻止免疫耐受并触发过度的炎性免疫反应[8]。
Rees等[9]实验证实,病原体入侵诱导IECs细胞应激,激活促炎性DC,增加细胞膜表面CD80、CD86、MHCⅡ类分子的表达及炎性因子IL-6、IL-15和IL-12的分泌,而减少表面标记CD103和抑炎性细胞因子IL-10的表达。Hart等[10]同样发现,呈激活状态的DC表面的模式识别受体[Toll样受体(toll-like receptor,TLR)2和TLR4]、MHCⅡ类分子以及共刺激分子(CD40和CD86)等表达上调,与健康对照组相比,可产生更多的炎性细胞因子,如IL-6、IL-12和肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)。克罗恩病(Crohn′s disease,CD)采用抗TNF-α单克隆抗体治疗后,DC共刺激分子的表达降低,表明在CD患者中DC促炎性表型的优势表达。Matsuno等[11]研究发现,溃疡性结肠炎(ulcerative colitis,UC)患者炎性黏膜中DC的数量增加,但CD103+DC亚群减少,且这些CD103+DC合成Treg的能力受损。因此,Treg合成减少以及促炎性Th1/Th17等效应T细胞的生成增多可能是IBD后续炎症反应放大的主要起因或延续因素。
IBD患者的DC失调并不局限于胃肠道黏膜,循环DC也表现出表型和功能的改变[12]。在健康状况下,血液DC表现出多种组织归巢的能力(如肠道、皮肤)[13]。但在IBD中,在肠道微环境及局部视黄酸刺激下,DC具有肠道特异性,表达黏膜归巢标记ɑ4β7和(或)回肠归巢标记CCR9[14],见图1。尽管如此,循环DC表型改变的机制目前尚不明确。可能是因为血浆中的促炎症介质在DC到达肠道靶组织之前就会调节其性质。由此可见,IBD中过度免疫炎症反应的发生与DC的数量及功能改变有关。然而,在IBD中DC数量及功能是如何被改变,进而导致过度炎症反应的具体机制尚不明确,亟待进一步探讨。
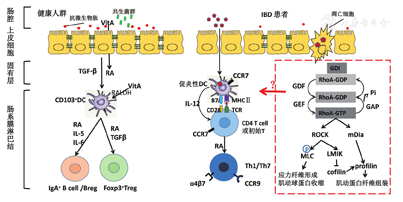
RhoA属于RhoGTPase亚家族,是Ras同源家族中的一类小鸟苷三磷酸酶。与Ras一样,Rho蛋白也是细胞外刺激介导信号网络的关键调节因子,目前在哺乳动物细胞中已发现22种Rho蛋白,其中RhoA、Rac1和Cdc42是研究最广泛的。RhoA是一种普遍表达的小GTP结合蛋白,在激活的情况下,RhoA与下游效应因子如Rho相关蛋白激酶(ROCK)和mDia1等相互作用。ROCK以磷酸化依赖性方式抑制肌球蛋白Ⅱ调节轻链磷酸酶(myosin Ⅱ regulatory light chain phosphatas,MLCP)活性,使磷酸化水平升高,从而导致活性MLC介导肌动球蛋白装配,诱导应力纤维和粘着斑的形成[15]。此外,ROCK也促使下游LIM域激酶(LIMK)的磷酸化依赖性激活,进而抑制下游肌动蛋白解离因子cofilin的活性[16]。活性RhoA还结合mDia1进而促使介导肌动蛋白聚合的profilin与肌动蛋白发生结合[17]。RhoA与下游效应因子结合并产生相应的生物学效应,这一系列反应构成RhoA信号通路,在调节细胞骨架蛋白、细胞形态和迁移以及细胞增殖和转录活性等方面发挥多种功能[18]。
DC在外周组织中的迁移具有两个主要功能,包括未成熟DC的抗原采样以及趋化因子引导的成熟DC向淋巴管和淋巴结迁移进行抗原递呈。这些迁移事件决定适应性免疫反应的效率。Vargas等[19]研究发现,DC的迁移取决于两个主要的肌动蛋白池,即位于DC前部的Cdc42-Arp2/3依赖性肌动蛋白池(促进抗原摄取但限制迁移)和后部的RhoA-mDia1依赖性肌动蛋白池(有助于细胞向前运动)。DC成熟后,前部的Arp2/3依赖性F-肌动蛋白富集明显减少,成熟DC切换为更快和更持久的mDia1依赖性运动模式,该模式促进DC趋化性迁移至淋巴管和淋巴结[19]。实验证实,Cdc42基因敲除小鼠或使用Cdc42抑制剂ML141产生的DC表现出更快的迁移速度,且抑制Cdc42的活性可显著降低DC的胞吞作用。相反,RhoA基因敲除或RhoA抑制的DC表现出迁移速度的降低,并且在用脂多糖处理时,DC在其前部积累F-肌动蛋白,通过胞吞作用摄取抗原。但是,DC的成熟不受Cdc42和RhoA缺陷的影响[19]。因此,成熟DC表现出RhoA-mDia1依赖性肌动蛋白聚集的快速迁移表型,而抑制RhoA活性则不利于DC的迁移从而影响抗原提呈。
DC是CD4+ T细胞最有效的激活剂,Kobayashi等[20]使用肉毒梭菌的外切酶C3作为RhoA抑制剂以及Y-27632作为特定的ROCK抑制剂(使p160ROCK失活)来探索RhoA/ROCK信号通路对DC及T细胞的影响;结果发现,采用C3及Y-27632干预后的DC形态和数量发生巨大的变化,表现为树突丢失、细胞膜的收缩和肌动蛋白聚合的显著减少;与CD4+ T细胞相关的DC数量比经C3处理的DC多2倍;通过电镜观察,未干预的DC与CD4 + T细胞的相互作用较经干预的DC更有效,干预后DC的同种抗原呈递能力以剂量依赖性方式被显著抑制;在同种异体混合淋巴细胞反应中,经C3干预的DC对T细胞的刺激能力降低了80%,这可能是由于DC与T细胞的初始相互作用被干扰。
Segain等[21]实验证实,在CD患者和2,4,6-三硝基苯磺酸诱发的UC大鼠的病变肠黏膜中,RhoA的活性增强;体外单独激活RhoA足以诱导TNF的产生,大鼠口服ROCK抑制剂Y-27632可显著减少结肠炎症反应,且Y-27632可抑制NF-κB活化以及核因子κB磷酸化激活。由此可以推测特异性抑制RhoA/ROCK信号通路能够预防肠道炎症的发生。
但是,也有研究发现激活RhoA信号通路反而能够减轻炎症反应的发生。Seul等[22]实验发现,在转基因小鼠中过表达p190 RhoGEF基因(一种RhoA活化基因),可负向调控DC对细菌脂多糖的反应,使得T细胞反应区缺乏活化的DC,血清IL-6水平降低;这些小鼠的DC中CD86、CD40和CD205的表达降低,但MHCⅡ类分子未见降低,同时对抗原摄取能力降低。可见RhoA的激活抑制了DC活化进而影响DC诱导的炎症反应。此外,MHCⅡ类分子的细胞定位也是DC依赖性炎性免疫应答的一个关键要素。有学者发现,SWAP-70是RhoA的抑制剂和RhoB活性的间接抑制剂,激活的SWAP-70-/-DC无法将MHCⅡ类分子正确定位在细胞膜,T细胞激活功能受到严重损害,并且F-肌动蛋白的重排被改变[23]。因此,MHCⅡ类分子的表面定位和T细胞活化可通过阻断RhoA和RhoB来恢复。ROCK抑制剂的使用同样可增加MHCⅡ类的膜表达及T细胞的激活[17]。Myo9B是一种RhoA活化抑制因子,Xu等[24]在Myo9B-/-DC中发现DC定向迁移能力及T细胞刺激能力受损,而Myo9B-/-DC受损的迁移和T细胞刺激能力很大程度上可以通过下游肌动蛋白解离因子cofilin的药理活化而恢复。这些研究都显示RhoA激活的DC表现出较差的迁移及激活T细胞的能力,进而可推测RhoA信号通路的激活可抑制炎症反应发生。然而这些实验都只证实了RhoA激活对DC功能及T细胞激活的抑制作用,而对于IBD的影响缺乏有力的数据支持,目前相关研究及报道仍较少。关于RhoA信号通路的改变对DC的影响以及进一步对IBD发生发展的意义还有待更详尽的研究。
DC在胃肠道黏膜免疫系统中具有重要作用,对实现免疫耐受并维持肠道稳态不可或缺。然而在病理情况下,DC也参与引发对内源性菌群或病原体的炎性T细胞应答,这是IBD的病理基础。DC除了决定适应性免疫反应的结果(致耐受性或促炎性)外,还确定淋巴细胞组织归巢的部位。而作为重要的细胞内信号分子开关,RhoA与细胞极性、细胞运动、细胞形状、细胞-细胞相互作用的调节以及胞吞和胞吐的调节密切相关。RhoA及其下游效应子ROCK和mDia1的变化同样影响DC的形态、数量及功能,进而可能影响DC的免疫反应及IBD的发生发展。鉴于DC作为固有免疫和适应性免疫应答之间桥梁的独特性质,以及RhoA信号通路对DC的重要影响,针对DC和RhoA信号通路相关分子机制及细胞功能的研究可以为IBD患者的临床诊断、监测、干预提供新的方向。但仍有一些问题尚待解决:首先,尽管CD和UC患者的黏膜DC表现出许多共同特征,但两者病理之间仍存在一些差异,其特异性机制仍然不明确。其次,IBD患者未受累黏膜中DC功能不同于炎性黏膜中DC功能的原因,以及IBD未受累黏膜与健康者黏膜DC间是否有差别等尚不清楚。此外,RhoA信号通路的改变对DC功能的影响是单一作用(抑制或激活)还是复杂作用机制下的某种作用优势表达,目前尚不明确。深入探讨DC和RhoA信号通路中有关IBD的发病机制,寻找新的生物治疗靶点,对治疗疾病有着十分重要的临床意义。
所有作者均声明不存在利益冲突

























