
Cronkhite-Canada综合征(CCS)是一种罕见疾病,以弥漫性胃肠道息肉、毛发及指甲脱落和皮肤色素沉着为临床特征,发病机制不明。本文报道1例CCS患者,经糖皮质激素治疗有效,基因检测发现结肠腺瘤样息肉基因突变,以期为临床诊治提供参考。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者男,70岁,因"间断腹泻4年,加重伴指甲萎缩、脱发、皮肤色素沉着8个月"于2019年6月28日入山西医科大学第一医院消化内科。2015年9月患者无明显诱因出现腹泻,5~6次/d,黄色稀水样,无腹痛、发热,并逐渐出现食欲减退、味觉减退,手指及脚趾指甲萎缩,毛发脱落,双手手掌及大腿内侧弥漫性棕褐色色素沉着,外院胃镜提示胃多发息肉,肠镜提示结肠多发息肉,口服美沙拉嗪肠溶片1 g tid,静脉使用地塞米松2.5 mg/d,共7 d,患者食欲较前改善,大便次数减少,后续口服泼尼松15 mg/d,共4 d后停药。患者手指及脚趾指甲新生,毛发再生,双手、双大腿内侧皮肤色素减退,期间仍有间断腹泻。2018年11月再次出现上述症状,外院复查胃镜提示胃角、胃窦、十二指肠黏膜病变待诊,肠镜提示结肠多发息肉,经内镜下息肉切除等对症治疗,疗效欠佳。自发病以来,精神欠佳,小便正常,1个月内体质量下降约7 kg。
入院后诊断:Cronkhite-Canada综合征(息肉-色素沉着-脱发-爪甲营养不良综合征,CCS)。查体:精神萎靡,神清,头发稀疏,手指及脚趾指甲萎缩,双手掌及足底色素沉着(图1),心肺腹无阳性体征。
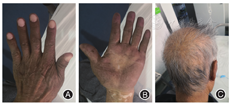
实验室检查:粪便常规+潜血正常,粪便培养阴性。血红蛋白129 g/L,白蛋白31.2 g/L,血钾3.03 mmol/L,C-反应蛋白为21.6 mg/L,红细胞沉降率35 mm/1 h,抗核抗体(ANA)及可提取的核抗原(ENA)抗体阴性,免疫球蛋白IgE略高。肿瘤标志物癌胚抗原为6.9 ng/ml,乙肝表面抗体、核心抗体和e抗体阳性,乙肝DNA提示病毒无复制,梅毒螺旋体、丙肝及艾滋病病毒抗体阴性,结核感染T细胞斑点试验(T-SPOT)阴性,血清EB-DNA及巨细胞病毒(CMV)-DNA阴性。
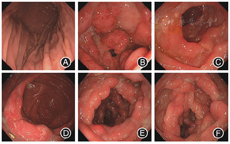
影像及内镜检查:胸部CT提示双肺散在实性小结节及陈旧条索影。腹部超声提示肝内钙化灶,双肾囊肿。胃镜提示胃底、胃体未见明显异常,胃窦及十二指肠降部多发息肉样隆起(图2A,图2B,图2C);肠镜提示结肠多发息肉(图2D,图2E,图2F)。
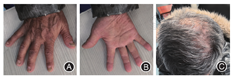
治疗经过:口服泼尼松25 mg/d,逐渐减量至停药(共半年),服用泼尼松1周后,患者食欲明显改善,未再腹泻,1个月后双手及脚趾指甲逐渐生长,手掌色素沉着明显减轻,头发生长,见图3。半年体质量增加5 kg。2020年1月10日复查肠镜:回盲部结节状息肉样肿物并融合,部分绒毛状,见图4;病理提示慢性炎细胞浸润。
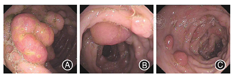
经过泼尼松口服治疗,患者症状明显改善,但结肠息肉有进展趋势,与患者及家属充分沟通后完善肿瘤相关基因检测(上海至本医学检验所),标本为结肠镜活检组织,测序结果:(1)肿瘤体细胞变异:651个基因的全部外显子,65个基因的部分内含子,发现结肠腺瘤样息肉(APC)等肿瘤相关基因变异7个,见表1;(2)对美国国立综合癌症网络(NCCN)指南提及的63个肿瘤遗传易感基因进行检测,未发现明确有害的可遗传胚系变异。
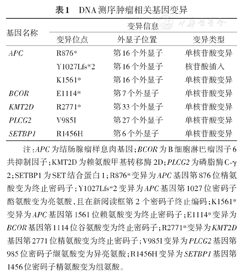
DNA测序肿瘤相关基因变异
DNA测序肿瘤相关基因变异
| 基因名称 | 变异信息 | ||
|---|---|---|---|
| 变异位点 | 外显子位置 | 变异类型 | |
| APC | R876* | 第16个外显子 | 单核苷酸变异 |
| Y1027Lfs*2 | 第16个外显子 | 核苷酸插入 | |
| K1561* | 第16个外显子 | 单核苷酸变异 | |
| BCOR | E1114* | 第7个外显子 | 单核苷酸变异 |
| KMT2D | R2771* | 第33个外显子 | 单核苷酸变异 |
| PLCG2 | V985I | 第27个外显子 | 单核苷酸变异 |
| SETBP1 | R1456H | 第6个外显子 | 单核苷酸变异 |
注:APC为结肠腺瘤样息肉基因;BCOR为B细胞淋巴瘤因子6共抑制因子;KMT2D为赖氨酸甲基转移酶2D;PLCG2为磷脂酶C-γ2;SETBP1为SET结合蛋白1;R876*变异为APC基因第876位精氨酸变为终止密码子;Y1027Lfs*2变异为APC基因第1027位密码子酪氨酸变为亮氨酸,且在新阅读框第2个密码子终止编码;K1561*变异为APC基因第1561位赖氨酸变为终止密码子;E1114*变异为BCOR基因第1114位谷氨酸变为终止密码子;R2771*变异为KMT2D基因第2771位精氨酸变为终止密码子;V985I变异为PLCG2基因第985位密码子缬氨酸变为异亮氨酸;R1456H变异为SETBP1基因第1456位密码子精氨酸变为组氨酸。
CCS于1955年首次报道[1],是一种罕见的以皮肤外胚层异常和胃肠道多发息肉为特征的综合征,但其发病机制尚不明确,部分CCS患者血浆IgG4水平升高[2],息肉IgG4阳性浆细胞浸润[3]、对免疫抑制治疗的反应[2]均提示可能和自身免疫相关。通过PubMed、中国知网以及万方医学数据库的检索,发现CCS的研究以个案报道居多[4,5],目前为止最多样本量的队列研究为日本学者对全国983家教学医院基于问卷的方式调查,回顾性分析210例CCS患者的临床特征、内镜检查结果、治疗方法以及长短期预后[6]。
关于CCS的临床表现,主要包括反复发作的腹泻、味觉减退、胃肠道多发息肉、脱发、甲营养不良、皮肤色素沉着,外胚层改变较为典型,同时结合胃肠道多发息肉,可以明确诊断CCS。
我们推测CCS可分为活动期和缓解期,活动期除有腹泻、胃肠道息肉的表现之外,最主要是合并外胚层改变,如毛发脱失、色素沉着、甲营养不良,经过糖皮质激素诱导缓解,疾病可进入缓解期,外胚层症状改善,部分患者胃肠道息肉明显减少甚至消失。本例患者经过口服泼尼松治疗后,腹泻症状消失,半年体质量增加5 kg,但是胃肠道息肉有进展,考虑是否与激素用量不足有关,亦或与患者本身APC基因突变有关。目前指南并没有提及CCS基因检测的必要性[7,8]。此患者发现APC基因突变是否偶然尚不确定。一项包括83例CCS患者的临床分析发现,CCS的胃肠道息肉组织病理类型以增生性息肉和腺瘤性息肉最为常见(71%),错构瘤性息肉较少(10%)[9]。CCS患者合并消化道恶性肿瘤的概率为10%~20%,早期发现的结肠癌主要位于腺瘤附近,癌变的发生与息肉的病理类型可能有关[6]。根据CCS的息肉病理类型,其癌变的发生可能遵循炎-癌或者腺瘤-癌变的演变顺序。
糖皮质激素是CCS的首选治疗方案,可诱导临床症状改善,且治疗后内镜下病变可减轻或消失,其他措施也以免疫调节为主,包括美沙拉嗪[10]、免疫抑制剂[11]、抗肿瘤坏死因子(tumor necrosis factor,TNF)-α单抗[12]。但关于糖皮质激素的剂量以及疗程尚无共识,刘颖娴等[11]对12例CCS患者的诊治过程及预后分析,提出泼尼松大于40 mg/d能使CCS患者获益更多,且激素治疗有效时应继续原剂量维持一段时间后再逐渐减量,总疗程最好大于6个月,以巩固疗效、降低复发率。
早期文献提示CCS预后不良,5年病死率为55%[13]。近年来发现以糖皮质激素为基础的治疗方案有助于延缓病情进展,Liu等[14]分析31例CCS患者的长期预后及内镜下表现,发现5年生存率为87.4%,较前明显提高,并且提出较大的胃息肉(最大直径为2 cm)是预后较差的预测因素,可能与肠套叠、胃肠道出血和恶性肿瘤的风险较高有关,但是结肠息肉的大小对预后没有明显影响。有报道CCS患者合并结直肠癌[15,16],这提示我们,即使糖皮质激素治疗有效,仍需要规律的内镜防癌筛查。
所有作者均声明不存在利益冲突

























