
探究合并艰难梭菌感染(CDI)的炎症性肠病(IBD)患者的肠道菌群特征。
采用横断面研究方法,连续纳入2017年11月至2018年4月在北京协和医院就诊的IBD患者54例。根据是否合并CDI,将患者分为CDI组(n = 20)和非CDI组(n = 34)。同时设健康对照40例。采用16S rRNA测序技术进行肠道菌群的测定,比较3组样本肠道微生物多样性和物种组成的差异。
与健康对照组相比,非CDI组和CDI组α多样性(Chao1指数、Shannon指数、Simpson指数、Pielou指数)更低(均P<0.05)。两组与健康对照组在β多样性(肠道菌群结构)的差异均存在统计学意义(均P<0.05)。CDI组和非CDI组间α多样性及β多样性差异均无统计学意义(均P>0.05)。与健康对照组相比,非CDI组和CDI组中粪杆菌属相对丰度更低,而变形菌门和肠杆菌科相对丰度更高,差异均具有统计学意义(均P<0.05)。与非CDI组相比,CDI组副拟杆菌属和罗斯氏菌属相对丰度更低,而拟梭菌属相对丰度更高,差异均具有统计学意义(均P<0.05)。Lefse分析显示活泼瘤胃球菌、梭菌属的无害梭菌和类腐败梭菌、香肠乳杆菌以及消化链球菌科是CDI组中对差异贡献显著的标志物种。
与健康人群及未合并CDI的IBD患者相比,IBD合并CDI患者肠道菌群物种组成存在特异性改变。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
炎症性肠病(inflammatory bowel disease,IBD)是一组由免疫介导的慢性炎症性消化道疾病,包括克罗恩病(Crohn′s disease,CD)和溃疡性结肠炎(ulcerative colitis,UC)。IBD患者多伴有肠道菌群失调和免疫紊乱,易发生艰难梭菌感染(Clostridium difficile infection,CDI)[1]。国内IBD患者CDI的发生率为5.0% ~ 15.4%[2]。艰难梭菌是一种可形成孢子、产毒的厌氧型芽孢杆菌,是寄生于人和动物胃肠道的机会性致病菌,也是引起医院获得性感染性腹泻的主要原因[3]。由于CDI可使IBD的病程复杂化,并可能导致不良预后[4],合并CDI的IBD越来越受到重视。但目前多数研究集中在IBD合并CDI的诊断、危险因素及治疗方面,如能总结其肠道菌群特征,探讨肠道微生态在这类患者中的可能作用,就可以为新治疗策略的开发指明方向。本研究旨在评估合并CDI的IBD患者肠道菌群特征,从肠道菌群的角度进一步阐明IBD与CDI的相互作用,为IBD合并CDI的诊治提供理论依据。
采用横断面研究方法,连续纳入2017年11月至2018年4月北京协和医院收治的54例IBD患者。纳入标准:(1)根据《炎症性肠病诊断与治疗的共识意见(2018年,北京)》诊断标准诊断为IBD[5];(2)因病情活动收入院并接受CDI筛查。排除标准:(1)入院前1个月内曾使用甲硝唑和(或)万古霉素;(2)曾接受粪菌移植治疗;(3)曾接受肠切除术和(或)肠造口术。
依据是否符合CDI诊断标准,将IBD患者分为CDI组及非CDI组。CDI定义为临床符合腹泻诊断标准,同时采用ELISA法检验粪便标本中艰难梭菌毒素A(-)B(+)或A(+)B(+)(VIDAS®C. difficile panel检测试剂盒购自法国BioMérieux公司),或经实时PCR检测毒素B编码基因tcdB阳性。纳入同期健康体检者40例,设为健康对照组,该组人群身体健康,无胃肠道、感染性疾病,无其他慢性病。
本研究已通过中国医学科学院北京协和医院伦理委员会批准(批准号:JS-1494)。
收集所有参与者的人口学信息及临床信息,包括年龄、性别、诊断、蒙特利尔分型、疾病活动度及收集粪便标本时所用药物。
在患者开始抗生素治疗之前使用无菌容器收集2 g粪便标本,2 h内转运至-80℃冰箱保存。采用十六烷基三甲基溴化铵法(CTAB)提取粪便样本基因组DNA,检测其纯度和浓度。
引物对应区域包括16S V4区(515F和806R,鉴定细菌多样性)、18S V4区(528F和706R,鉴定真核微生物多样性)、ITS1区(ITS5-1737F和ITS2-2043R,鉴定真菌多样性)、细菌16S V3-V4/V4-V5区、古菌16S V4/V4-V8区、真核生物18S V9区和真菌ITS2区。引物设计和合成由诺禾致源公司完成。引物序列见表1。

引物序列
引物序列
| 类型 | 区域 | 引物序列(5 ′ →3 ′) |
|---|---|---|
| 细菌16S | V4 | 正向GTGCCAGCMGCCGCGGTAA |
| 反向GGACTACHVGGGTWTCTAAT | ||
| V3-V4 | 正向CCTAYGGGRBGCASCAG | |
| 反向GGACTACNNGGGTATCTAAT | ||
| V4-V5 | 正向GTGCCAGCMGCCGCGGTAA | |
| 反向CCGTCAATTCCTTTGAGTTT | ||
| 古菌16S | V4 | 正向CAGCCGCCGCGGTAA |
| 反向GTGCTCCCCCGCCAATTCCT | ||
| V4-V8 | 正向TTWAGTCAGGCAACGAGC | |
| 反向TGTGCAAGGAGCAGGGAC | ||
| 真核生物18S | V4 | 正向GCGGTAATTCCAGCTCCAA |
| 反向AATCCRAGAATTTCACCTCT | ||
| V9 | 正向CCCTGCCHTTTGTACACAC | |
| 反向CCTTCYGCAGGTTCACCTAC | ||
| 真菌ITS | ITS1 | 正向GGAAGTAAAAGTCGTAACAAGG |
| 反向GCTGCGTTCTTCATCGATGC | ||
| ITS2 | 正向GCATCGATGAAGAACGCAGC | |
| 反向TCCTCCGCTTATTGATATGC |
以基因组DNA为模板,使用带Barcode的特异引物、美国New England Biolabs公司的Phusion® High-Fidelity PCR Master Mix with GC Buffer和高效高保真酶进行PCR。PCR反应体系:Phusion Master Mix(2×)15 μl,正反向引物各1 μl,gDNA(1 ng/μl)10 μl,ddH2O补足至30 μl。反应程序:98℃预变性1 min;98℃变性10 s,50℃退火30 s,72℃延伸30 s,共30个循环;72℃持续5 min使反应产物扩增充分。
PCR产物使用2%浓度的琼脂糖凝胶进行电泳检测并纯化,验证完整性后回收目标条带。使用美国Thermofisher Scientific公司Ion Plus Fragment Library Kit 48 rxns建库试剂盒进行文库构建,然后经Qubit定量和文库检测合格后,使用Thermofisher Scientific公司的Ion S5TMXL进行上机测序。
使用Cutadapt(v1.9.1)软件对测序数据初步处理得到原始数据,再通过UCHIME Algorithm将原始数据与物种注释数据库进行比对并去除嵌合体序列,得到有效数据。使用Uparse(v7.0.1001)软件将所有有效数据以97%的一致性聚类成为操作分类单元(operational taxonomic units,OTUs)并选取代表序列,用Mothur方法与SILVA的SSUrRNA数据库进行物种注释分析,统计各样本群落组成,对各样品的数据进行均一化处理。使用QIIME软件对均一化处理后的OTUs进行α多样性分析(Chao1、Shannon、Simpson、Pielou指数)和β多样性分析,计算Unifrac距离。Unifrac距离是利用各样本中微生物序列间的进化信息计算的样本间距离,反映不同样本间的微生物群落构成即β多样性。使用R(v4.0.3)WGCNA、stats和ggplot2软件包进行主坐标分析(principal coordinates analysis,PCoA)。通过Anosim分析判断组间群落结构差异是否有统计学意义。Lefse分析是一种将秩和检验和线性判别分析(linear discriminant analysis,LDA)相结合的分析方法,强调统计意义和生物相关性,可用于寻找不同组间的生物标志物,LDA score反映了各标志物对组间差异的影响和贡献程度。本研究通过Lefse(LDA effect size)分析寻找在组间差异具有统计学意义的生物标志物,设定LDA score阈值为2。
采用R(v4.0.3)软件,正态分布的连续变量以 ± s表示,组间比较采用t检验(2组)或单因素方差分析(3组);偏态分布的连续变量以[M(Q1,Q3)]表示,组间比较采用Mann-Whitney U检验(2组)或Kruskal-Wallis检验(3组)。分类变量以百分比表示,2组间比较采用卡方检验或Fisher精确检验,3组间比较采用R×C列联表卡方检验。多重比较采用Benjamini-Hochberg法校正P值,以P<0.05或多重校正后错误发现率(false discovery rate,FDR)<0.05为差异具有统计学意义,结果中多重比较的P值均为校正后P值。Anosim检验中,R值介于-1 ~ 1,若R>0且P<0.05,说明组间差异具有统计学意义。
± s表示,组间比较采用t检验(2组)或单因素方差分析(3组);偏态分布的连续变量以[M(Q1,Q3)]表示,组间比较采用Mann-Whitney U检验(2组)或Kruskal-Wallis检验(3组)。分类变量以百分比表示,2组间比较采用卡方检验或Fisher精确检验,3组间比较采用R×C列联表卡方检验。多重比较采用Benjamini-Hochberg法校正P值,以P<0.05或多重校正后错误发现率(false discovery rate,FDR)<0.05为差异具有统计学意义,结果中多重比较的P值均为校正后P值。Anosim检验中,R值介于-1 ~ 1,若R>0且P<0.05,说明组间差异具有统计学意义。
共纳入54例IBD患者,非CDI组34例,CDI组20例。非CDI组与CDI组在年龄、性别、疾病类型、蒙特利尔分型、严重程度和药物史方面的差异均无统计学意义(均P>0.05),见表2。共纳入健康对照者40例,年龄(24.2 ± 2.2)岁,其中男性12例,女性28例。
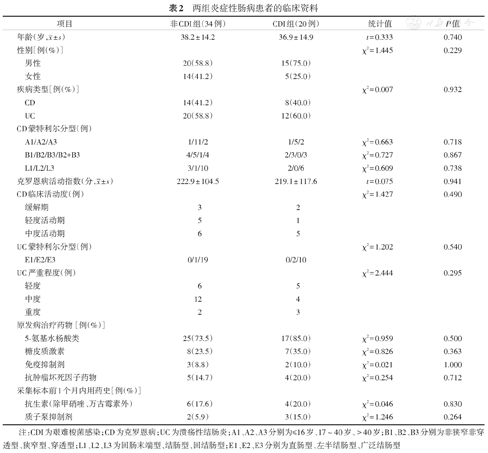
两组炎症性肠病患者的临床资料
两组炎症性肠病患者的临床资料
| 项目 | 非CDI组(34例) | CDI组(20例) | 统计值 | P值 | |
|---|---|---|---|---|---|
年龄(岁, ± s) ± s) | 38.2 ± 14.2 | 36.9 ± 14.9 | t = 0.333 | 0.740 | |
| 性别[例(%)] | χ2=1.445 | 0.229 | |||
| 男性 | 20(58.8) | 15(75.0) | |||
| 女性 | 14(41.2) | 5(25.0) | |||
| 疾病类型[例(%)] | χ2=0.007 | 0.932 | |||
| CD | 14(41.2) | 8(40.0) | |||
| UC | 20(58.8) | 12(60.0) | |||
| CD蒙特利尔分型(例) | |||||
| A1/A2/A3 | 1/11/2 | 1/5/2 | χ2=0.663 | 0.718 | |
| B1/B2/B3/B2+B3 | 4/5/1/4 | 2/3/0/3 | χ2=0.727 | 0.867 | |
| L1/L2/L3 | 3/1/10 | 2/0/6 | χ2=0.609 | 0.738 | |
克罗恩病活动指数(分, ± s) ± s) | 222.9 ± 104.5 | 219.1 ± 117.6 | t = 0.075 | 0.941 | |
| CD临床活动度(例) | χ2=1.427 | 0.490 | |||
| 缓解期 | 3 | 2 | |||
| 轻度活动期 | 5 | 1 | |||
| 中度活动期 | 6 | 5 | |||
| UC蒙特利尔分型(例) | χ2=1.202 | 0.540 | |||
| E1/E2/E3 | 0/1/19 | 0/2/10 | |||
| UC严重程度(例) | χ2=2.444 | 0.295 | |||
| 轻度 | 6 | 5 | |||
| 中度 | 12 | 4 | |||
| 重度 | 2 | 3 | |||
| 原发病治疗药物[例(%)] | |||||
| 5-氨基水杨酸类 | 25(73.5) | 17(85.0) | χ2=0.959 | 0.500 | |
| 糖皮质激素 | 8(23.5) | 7(35.0) | χ2=0.826 | 0.363 | |
| 免疫抑制剂 | 3(8.8) | 2(10.0) | χ2=0.021 | 1.000 | |
| 抗肿瘤坏死因子药物 | 5(14.7) | 4(20.0) | χ2=0.254 | 0.712 | |
| 采集标本前1个月内用药史[例(%)] | |||||
| 抗生素(除甲硝唑、万古霉素外) | 6(17.6) | 4(20.0) | χ2=0.046 | 0.830 | |
| 质子泵抑制剂 | 2(5.9) | 3(15.0) | χ2=1.246 | 0.264 | |
注:CDI为艰难梭菌感染;CD为克罗恩病;UC为溃疡性结肠炎;A1、A2、A3分别为≤16岁、17 ~ 40岁、>40岁;B1、B2、B3分别为非狭窄非穿透型、狭窄型、穿透型;L1、L2、L3为回肠末端型、结肠型、回结肠型;E1、E2、E3分别为直肠型、左半结肠型、广泛结肠型
3组共94个样本上机测序,数据经初步质控、去除barcode、primer和嵌合体后,共获得7 295 235条高质量序列,平均每个样本77 609条序列(序列范围43 620 ~ 91 692)。以97%的一致性聚类共获得3186个OTUs,其中CDI组1988个,非CDI组2074个,健康对照组2729个。各样本稀释曲线均接近饱和,表明测序数据量合理、深度达到要求;物种累积箱型图随样本量加大趋于平缓,表明抽样充分可进行数据分析。
通过计算α多样性指数分析3组样本的物种丰富度(Chao1、Shannon指数)和均匀度(Simpson、Pielou指数),结果显示,3组间上述4项指标总体差异均有统计学意义(均P<0.001);多重比较中,非CDI组和CDI组上述4项指标均显著低于健康对照组,差异均具有统计学意义(均P<0.05);非CDI组和CDI组间4项指标差异均无统计学意义(均P>0.05)。见表3。
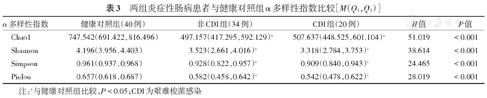
两组炎症性肠病患者与健康对照组α多样性指数比较[M(Q1,Q3)]
两组炎症性肠病患者与健康对照组α多样性指数比较[M(Q1,Q3)]
| α多样性指数 | 健康对照组(40例) | 非CDI组(34例) | CDI组(20例) | H值 | P值 |
|---|---|---|---|---|---|
| Chao1 | 747.542(691.422,816.496) | 497.157(417.295,592.129)a | 507.637(448.525,601.104)a | 51.019 | <0.001 |
| Shannon | 4.196(3.956,4.403) | 3.523(2.661,4.016)a | 3.318(2.784,3.753)a | 38.614 | <0.001 |
| Simpson | 0.961(0.937,0.968) | 0.928(0.822,0.957)a | 0.909(0.840,0.943)a | 24.465 | <0.001 |
| Pielou | 0.657(0.618,0.687) | 0.582(0.458,0.642)a | 0.542(0.478,0.622)a | 28.019 | <0.001 |
注:a与健康对照组比较,P<0.05;CDI为艰难梭菌感染
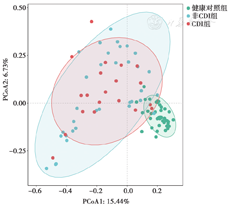
通过PCoA分析对样本进行聚类,PCoA1、PCoA2分别为第一、第二主坐标对样本矩阵差异的贡献度。本组PCoA1为15.44%,PCoA2为6.73%,结果显示非CDI组和CDI组均与健康对照组明显分离。经Anosim检验,2组IBD患者与健康对照组的肠道菌群结构差异均具有统计学意义(CDI组比健康对照组:R = 0.674,P = 0.0015;非CDI组比健康对照组:R = 0.438,P = 0.0015)。而非CDI组与CDI组未明显分离,Anosim检验示非CDI组和CDI组间肠道微生物群落结构差异无统计学意义(R = -0.0058,P = 0.509)。见图1。
门水平上,3组的优势菌门均为厚壁菌门、拟杆菌门、变形菌门和放线菌门。其中,3组间厚壁菌门、变形菌门和放线菌门相对丰度的差异均具有统计学意义(均P<0.05)。多重比较中,非CDI组和CDI组中变形菌门相对丰度均高于健康对照组,非CDI组厚壁菌门相对丰度则低于健康对照组(均P<0.05)。在变形菌门中,3组间肠杆菌科的相对丰度差异具有统计学意义(P = 0.001),并且非CDI组和CDI组均高于健康对照组(均P<0.05)。见表4。
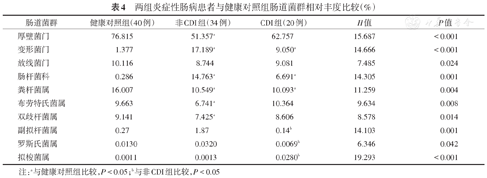
两组炎症性肠病患者与健康对照组肠道菌群相对丰度比较(%)
两组炎症性肠病患者与健康对照组肠道菌群相对丰度比较(%)
| 肠道菌群 | 健康对照组(40例) | 非CDI组(34例) | CDI组(20例) | H值 | P值 |
|---|---|---|---|---|---|
| 厚壁菌门 | 76.815 | 51.357a | 62.757 | 15.687 | <0.001 |
| 变形菌门 | 1.377 | 17.189a | 9.050a | 14.666 | <0.001 |
| 放线菌门 | 10.116 | 8.744 | 9.081 | 7.485 | 0.024 |
| 肠杆菌科 | 0.286 | 14.763a | 6.691a | 14.305 | 0.001 |
| 粪杆菌属 | 16.007 | 10.549a | 10.093a | 11.259 | 0.004 |
| 布劳特氏菌属 | 9.663 | 6.741a | 10.364 | 9.634 | 0.008 |
| 双歧杆菌属 | 9.141 | 7.425a | 8.606 | 8.578 | 0.014 |
| 副拟杆菌属 | 0.27 | 1.87 | 0.14b | 14.103 | 0.001 |
| 罗斯氏菌属 | 0.0130 | 0.0320 | 0.0069b | 6.346 | 0.042 |
| 拟梭菌属 | 0.0011 | 0.0013 | 0.0280b | 19.293 | <0.001 |
注:a与健康对照组比较,P<0.05;b与非CDI组比较,P<0.05
属水平上,3组样本优势菌属为拟杆菌属、粪杆菌属、布劳特氏菌属和双歧杆菌属。其中,3组间共有98种菌属的相对丰度差异具有统计学意义,优势菌属中的粪杆菌属、布劳特氏菌属和双歧杆菌属相对丰度的差异均具有统计学意义(均P<0.05)。在多重比较中,非CDI组和CDI组粪杆菌属相对丰度均低于健康对照组,非CDI组中布劳特氏菌属和双歧杆菌属相对丰度均低于健康对照组(均P<0.05)。与非CDI组相比,CDI组样本中副拟杆菌属(0.14%比1.87%,Z = 2.634,P = 0.017)和罗斯氏菌属(0.0069%比0.0320%,Z = 2.512,P = 0.036)的相对丰度更低,而拟梭菌属(0.0280%比0.0013%,Z = 4.254,P<0.001)的相对丰度更高。见表4。
将非CDI组与CDI组的物种注释结果进行Lefse分析,发现CDI组中对差异贡献显著的标志微生物单元包括活泼瘤胃球菌、梭菌属的无害梭菌和类腐败梭菌、香肠乳杆菌以及消化链球菌科;而非CDI组主要包括坦纳菌科、副拟杆菌属、链球菌属、拟杆菌属以及厚壁菌门的Faecalicoccus等,见图2。与非CDI组和CDI组相比,健康对照组对差异贡献显著的菌属共有28个,其中LDA score>4的有粪杆菌属、罗氏菌属、双歧杆菌属、纺锤链杆属和unidentified_Ruminococcaceae。
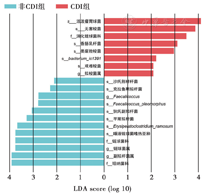
与健康人群相比,IBD患者存在显著的菌群失调和特征性菌群改变。既往研究指出,这些改变不仅是肠道炎症的结果,也可能是疾病发生的主要因素[6]。菌群组成、功能和代谢的改变可使肠道微生态营养失衡、肠道组织受累、炎症调节功能减弱,从而使得机会致病菌如艰难梭菌易于发生定植和感染[7]。CDI是IBD患者中常见的机会性感染,合并CDI是IBD患者病情加重和接受结肠切除术的危险因素[8,9]。因此,研究IBD合并CDI患者的肠道菌群改变可有助于更好地理解疾病发生发展的机制,也可为开发新的生物标志物和治疗方法提供参考。
本研究对样本进行多样性分析发现,与健康人群相比,IBD患者无论是否合并CDI,其肠道菌群的多样性和丰富度均更低,并且肠道菌群构成的差异存在统计学意义。但非CDI组和CDI组之间的α及β多样性差异并无显著统计学意义。这与文献报道欧美人群中的结果一致[10,11,12]。但也有学者提出不同看法,Bushman等[11]指出,是否合并CDI对IBD微生物组的影响极不均衡,难以产生显著的强分辨信号,同时2组患者药物暴露较为相似,可能以相似的方式影响群落的组成。Chen等[13]通过16S rRNA测序发现,与未合并艰难梭菌定植的IBD儿童相比,合并无症状艰难梭菌定植的IBD儿童患者中肠道菌群的α多样性更低,群落结构发生改变。因受样本量、患者人群特征、不同测序方法的影响,目前对于合并CDI的IBD患者肠道菌群整体多样性和群落结构的改变尚无一致结论,未来有待样本量更大、人群特征覆盖度更广的队列研究进一步探索。
本研究发现,合并CDI的IBD患者的优势菌群与健康对照组差异存在统计学意义。由Lefse分析可知,健康对照组中厚壁菌门以及厚壁菌门下粪杆菌属、罗氏菌属、Ruminococcaceae等类群较合并CDI的IBD患者更为富集。这些类群多为产丁酸盐菌,Ruminococcaceae则可消耗氢并生成醋酸盐,为罗氏菌属产生丁酸盐提供原料[14]。丁酸盐是维持肠道微生态平衡的关键代谢物,也是结肠上皮细胞的主要能量来源,同时具有免疫调节特性,在肠道发挥抗炎作用[15]。因此,这些微生物的丰度下降可能导致正常炎症调节能力下降、肠道黏膜表面氧化应激增加。此外,本研究发现合并CDI的IBD患者变形菌门以及变形菌门下肠杆菌科的丰度显著高于健康对照组。与梭菌亚群IV和XIVa相比,氧化应激环境对肠杆菌科的生长更为有利[15]。因此,其中的机会致病菌更易定植,并可通过诱导细胞因子的产生、破坏肠道稳态直接影响疾病进展[7]。以上物种组成结果与既往IBD患者肠道菌群研究结论一致[7,16,17]。但值得注意的是,一项针对IBD患者肠道微生物地域分布特征的研究指出,罗氏菌属丰度降低只特异性地见于欧洲IBD患者[18]。而本研究发现中国IBD患者的肠道菌群也有类似改变,提示罗氏菌属在中国人群中亦可能作为益生菌,成为中国IBD人群益生菌或粪菌移植治疗的选择。
与非CDI组相比,本研究发现活泼瘤胃球菌以及梭菌属是CDI组患者中相对丰度更高的标志类群,这与此前一项法国IBD合并CDI患者的菌群研究报道的结果一致[10]。在Lefse分析中,活泼瘤胃球菌是LDA score最高的类群,它是一种黏液降解菌,可使用肠道黏液层的黏蛋白单糖作为能量来源,消耗唾液酸并产生丙酸和丙醇[19],可破坏黏膜屏障,增加细菌渗透性,诱导多种促炎因子的产生[6,20]。此外,在CD患者中,活泼瘤胃球菌可合成并分泌一种具有鼠李糖骨架和葡萄糖侧链的复合葡萄糖多糖,这一多糖可诱导树突状细胞以toll样受体4依赖的形式分泌促炎性细胞因子如肿瘤坏死因子α[21]。另外,活泼瘤胃球菌本身导致的黏膜屏障破坏可能增加了免疫系统对于促炎性多糖的暴露和敏感性。因此,两种机制相互叠加,导致肠道炎症反应加剧。Fuentes等[22]发现粪菌移植治疗失败的UC患者所接受的粪菌供体中含有较高水平的活泼瘤胃球菌(>2%),与粪菌移植后获得持续缓解的UC患者所接受粪菌供体相比升高3.8倍。在IBD合并CDI患者中,丰度较高的活泼瘤胃球菌亦可能是促进炎症发生与进展的主要因素之一。
本研究存在一定局限性。首先,肠道菌群受性别、年龄、疾病分期、疾病类型、治疗药物等多种因素的影响,而本研究纳入的受试者样本量相对较小,未严格匹配对照组与研究组的性别和年龄,肠道菌群的结构和丰度分析易产生偏倚,同时也难以根据疾病类型或治疗药物等因素对IBD患者进行亚组分析。其次,本研究为横断面研究,仅能反映IBD患者合并CDI在抗生素治疗前这一时间节点的肠道菌群特征,而不能阐明艰难梭菌感染前后、抗生素治疗前后及IBD疾病不同时期的肠道菌群动态变化特点,也无法说明菌群改变与IBD患者发生CDI的因果关系。未来有待更大的样本量和纵向队列对以上问题进行探究。
总体而言,本研究表明,与健康人群及单纯IBD患者相比,IBD合并CDI患者存在特征性的肠道菌群改变,这些特征性改变有望为IBD合并CDI的治疗提供新的思路。
所有作者均声明不存在利益冲突

























