
探讨Dickkopf–1 (Dkk–1)在类风湿关节炎(RA)滑膜炎症的作用。
将Dkk–1特异性siRNA转染至分离培养的滑膜成纤维细胞(RASF)使Dkk–1基因沉默;将Dkk–1质粒转染至RASF使Dkk–1基因过表达。应用实时荧光定量PCR(Real–time PCR)及酶联免疫吸附测定法(ELISA),检测RASF在转染前后几种致炎因子在mRNA及蛋白水平的表达变化。
Dkk–1特异性siRNA转染后随着Dkk–1表达水平受到抑制,白细胞介素(IL)–1β、肿瘤坏死因子(TNF)–α、IL–6及IL–8的表达水平降低。Dkk–1质粒转染后随着Dkk–1基因过表达,上述致炎因子表达水平增高。更为重要的是,RASF在TNF–α和IL–1β活化后,上述效应仍较为显著。
Dkk–1促进RASF分泌多种致炎因子,加重RA关节局部滑膜炎症。抑制关节局部Dkk–1的表达可减轻RA关节滑膜炎症,因此DKK–1有可能成为RA生物治疗的新靶标。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
滑膜炎症、血管翳形成及骨质破坏是类风湿关节炎(RA)患者关节局部最主要的病理学特征。RA患者关节局部大量炎性细胞浸润,活化滑膜组织中CD4+T细胞、巨噬细胞,产生大量炎性因子,作用于滑膜成纤维细胞(RASF),引发炎症反应[1]。RASF长期处于炎症状态,进一步造成局部骨及关节下软骨组织的破坏[2]。Wnt信号通路是介导异常免疫反应与骨代谢系统之间相互作用的重要通路,而该通路中的抑制蛋白Dkk(Dickkopf)–1是近年来新发现的介导免疫炎症与骨破坏的重要桥梁分子[3]。研究发现,Dkk–1在RA外周血及滑膜组织中异常增多,并与反映病情轻重的临床指标密切相关[4]。本研究针对Dkk–1对RA滑膜炎症的影响,应用小干扰RNA(siRNA)基因敲低和质粒转染超表达的方法处理RASF,从正反两方面研究Dkk–1在RA局部滑膜炎中的作用机制及潜在治疗价值。
由2013年4至7月北京大学人民医院收治的关节置换的RA患者(年龄30~60岁)处收集到新鲜的滑膜组织,患者均符合2009年美国风湿病学会关于RA的诊断标准。分离及培养滑膜细胞均在无菌条件下进行。用PBS将滑膜组织冲洗数遍后,在DMEM培养基中(Invitrogen)中用剪刀将脂肪组织剔除,将滑膜组织剪成细小的组织块置于无菌培养皿中,加入4 g/L胶原酶(美国Sigma公司)37 ℃消化2 h。消化后,使用PBS洗涤2遍后,应用含10% FBS,1×105U/L青霉素及100 mg/L链霉素的DMEM培养液,在5% CO2,37 ℃条件下培养,待70%~80%的细胞融合时按1∶3的比例传代。实验用4~8代细胞。RASF的鉴定采用流式细胞术检测CD14、CD68及vimentin蛋白,98%以上细胞为CD14、CD68阴性,vimentin蛋白阳性,证实分离出的细胞为RASF。
应用化学法合成靶向Dkk–1siRNA。转染前将细胞以5×104/孔的密度接种到6孔板里,转染前保证70%~80%的细胞融合。按转染试剂(Santa Cruz)及siRNA说明书所示,每孔细胞中加入4 μl siRNA及转染试剂。24 h后用Real–time PCR及ELISA方法检测敲减效果。
转染前将细胞以1×105/孔密度接种到6孔板里,转染前保证70%~80%的细胞融合。按转染试剂(Santa Cruz)说明书,每孔细胞中加入2 μg Dkk–1质粒或空载体对照。通过Real–time PCR及ELISA方法检测转染效果。
RASF转染24 h后,活化组细胞中加入人重组TNF–α(10 μg/L)和IL–1β(2 μg/L)蛋白刺激。4 h后收集细胞,应用TRIzol(Invitrogen)依照使用说明提取总RNA。所有有效的RNA都使用M–MLV逆转录酶(Fermentas,美国)合成cDNA。应用SYBR green(Applied Biosystems)试剂盒在ABI–7300检测系统进行Real–time PCR检测RASF中Dkk–1、致炎因子肿瘤坏死因子(TNF)–α、白细胞介素(IL)–6、IL–8及IL–1β表达水平。扩增引物由上海生工生物工程公司合成。建立总体积20 μl反应体系,反应条件:95 ℃ 15 s,60 ℃ 1 min,共40个循环。每份标本做3复孔,复孔间Ct值差异控制在1以内。采用相对定量法,以2–ΔCt值表示目的基因mRNA表达水平的高低[5]。
RASF转染24 h后,活化组细胞中加入人重组TNF–α(10 μg/L)和IL–1β(2 μg/L)蛋白刺激,24 h后收集培养上清。培养上清液经离心,应用商品化的ELISA试剂盒(R&D Minneapolis,MN,美国),根据说明书方法检测上清液中Dkk–1水平;应用商品化的ELISA试剂盒(欣博盛)根据说明书方法检测上清中IL–6、IL–8水平。
采用SPSS 16.0统计软件,不同组间比较采用Wilcoxon signed–rank test,以P<0.05为差异有统计学意义。
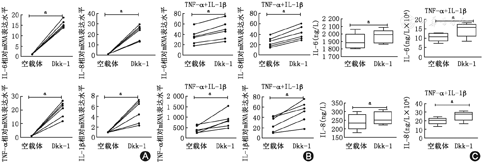
转染Dkk–1特异性siRNA24 h后,用Real time PCR及ELISA方法检测RASF中Dkk–1的表达水平(图1)。Real–time PCR结果示,Dkk–1特异性siRNA在mRNA水平有效抑制Dkk–1的表达,在TNF–α+IL–1β活化后的RASF中此抑制效果依然显著;ELISA方法检测培养上清中Dkk–1的表达水平结果显示Dkk–1特异性siRNA干扰后分泌到细胞培养上清中的Dkk–1水平明显下调,在TNFα+IL–1β活化后的RASF,此抑制效果仍然显著(P<0.05)。

转染Dkk–1特异性siRNA后,Real–time PCR的结果显示,RASF致炎因子IL–6、IL–8、TNF–α及IL–1β在转录水平表达量显著降低,即使在TNF–α和IL–1β联合刺激模拟RA关节局部炎性微环境后,仍能有效抑制上述致炎因子的表达(均P<0.05),结果分别见图2A及图2B。用ELISA的方法检测培养上清中IL–6及IL–8的表达水平,Dkk–1基因敲减后致炎因子IL–6和IL–8的表达受到明显抑制(均P<0.05),结果见图2C。上述结果表明,抑制RASF内源性Dkk–1的表达能一定程度上抑制致炎因子的释放,减轻滑膜炎症反应。

Dkk–1质粒转染24 h后,用Real–time PCR及ELISA方法检测RASF中Dkk–1的表达水平(结果见图3)。Dkk–1质粒转染后能明显增加Dkk–1表达水平。TNF–α和IL–1β联合刺激后,Dkk–1表达量进一步增高,提示RA关节局部的炎性环境能促进Dkk–1分泌增加(P<0.05)。
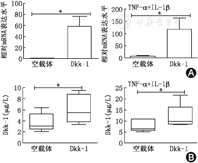
Dkk–1基因过表达后,RASF中致炎因子IL–6、IL–8、TNF–α及IL–1β在mRNA水平的表达量明显增高(图4A),此作用在TNF–α和IL–1β联合刺激模拟RA关节局部炎性微环境中进一步放大(图4B, P<0.05)。用ELISA的方法检测上清中IL–6及IL–8的表达水平,Dkk–1基因过表达后致炎因子IL–6和IL–8的表达显著增高(结果见图4C,P<0.05)。
Dkk(Dickkopf)–1是Glinka等[6]在1998年发现的一种蛋白,是Wnt信号通路的拮抗剂。Wnt信号通路促进间充质干细胞向成骨细胞分化。研究表明,Wnt介导的信号通路在RA骨质代谢的病理生理过程中起着关键性作用[7]。而Dkk–1与细胞膜上Wnt联合受体结合,从而抑制Wnt信号通路对成骨细胞增殖和分化的促进作用,抑制Dkk–1的表达可促进成骨细胞的分化。另外,成熟成骨细胞中的Wnt活化,可上调骨保护素(OPG)的表达,而OPG可抑制核因子κB受体活化因子配体(RANKL)诱导的破骨细胞分化作用,从而抑制骨侵蚀[8],近年来的研究提出,Dkk–1是联系RA免疫损伤与骨再塑的关键分子。在TNF–α等免疫炎症因子存在的条件下,Wnt–Dkk与OPG–RANKL两个系统相互作用直接影响成骨细胞与破骨细胞在关节组织中的平衡,导致破骨细胞异常活化,从而引起RA的骨质破坏。
近期研究显示[6,7,8,9],应用Dkk–1的单克隆抗体,能够使RA小鼠模型的骨破坏性模式转化为骨增生模式,而TNF–α抑制剂可使患者血清Dkk–1表达明显下调,说明Dkk–1在RA关节局部的高表达与局部炎性微环境密切相关。
研究显示,Dkk–1除了通过抑制Wnt通路直接作用于成骨和破骨细胞外,在RA患者滑膜细胞中也存在高水平表达[3],提示其可能与RASF活化增生及炎症有关。RASF是一种特殊的滑膜成纤维细胞。很多研究表明,其形态和生物学特性与普通滑膜成纤维细胞相比已发生了很大的变化。其在关节局部大量增殖并持续保持炎性状态。RASF在关节局部分泌大量致炎因子,如IL–6、IL–8、TNF–α等,为关节局部提供了炎性微环境,,这些致炎因子正向调节破骨细胞分化、软骨侵蚀,促进关节局部骨质破坏、血管翳形成及炎性免疫反应,在RA的关节破坏中发挥了重要作用[10]。
本研究发现,RASF表达的内源性Dkk–1能促进其分泌IL–6、IL–8、TNF–α及IL–1β,加重RA关节局部的滑膜炎症。RASF经过反复传代,在无诱导因子刺激的情况下可能出现返祖现象,分泌炎性因子的能力可能会下降[11]。所以本研究中在转染siRNA或质粒后予以TNF–α及IL–1β刺激RASF促使其恢复分泌炎性因子的能力,Dkk–1对上述致炎因子的促进作用进一步放大。而此种炎性微环境又能进一步刺激RASF内源性Dkk–1的表达,形成一种正反馈调节,加重RA关节局部的炎症反应,并促进骨质破坏。我们同时用不同浓度的重组人Dkk–1蛋白刺激RASF,却无法得到类似Dkk–1基因超表达造成内源性Dkk–1表达量增高促进炎性因子分泌的效果,在后续的研究中有必要对两种刺激方式之间差异的进一步研究和讨论。
另外一方面,通过构建Dkk–1特异性siRNA,减少RASF中内源性Dkk–1的表达,能有效抑制RASF分泌炎性因子,这种抑制效果在TNF–α、IL–1β活化后RASF中依然显著。这表明,通过siRNA抑制Dkk–1的表达能起到减轻RA关节局部滑膜炎症的作用。该方法可能成为RA治疗的新手段。

























