
探讨以间质性肺疾病(ILD)起病的皮肌炎患者的临床特点和预后。
北京协和医院1999年1月至2013年1月皮肌炎并ILD住院患者资料完整者共184例,回顾性分析其临床表现、生化指标、抗体谱、影像学、肺功能、肺活体组织检查学、治疗及转归等,并对ILD起病组及非ILD起病组皮肌炎患者进行上述指标的比较。
皮肌炎住院患者并发ILD的发生率为17%,平均年龄(48±12)岁,男∶女= 63∶121。ILD起病组88例(47.8%),包括ILD与皮肌炎同时发生(1个月内)42例(22.8%),以及ILD先于皮肌炎发生46例(25.0%),后者ILD平均先于皮肌炎(11±3)个月发生。ILD起病组活动后气短、咳嗽和肺部爆裂音的发生率高于非ILD起病组,而向阳疹、前胸V区皮疹、披肩征和关节受累的发生率较后者低(P< 0.05)。ILD起病组与非ILD起病组相比,肌酶水平、抗核抗体和抗SSA阳性率更低(P< 0.05),抗Jo–1抗体阳性率(13.6%)差异无统计学意义(P>0.05)。ILD起病的皮肌炎主要表现为弥散功能及限制性通气功能障碍,一氧化碳弥散率显著低于非ILD起病组(P< 0.01)。ILD起病的皮肌炎临床病理类型主要是机化性肺炎和非特异性间质性肺炎。ILD起病组病死率19.3%,两组差异无统计学意义(P> 0.05)。死亡原因主要是ILD进展所致呼吸衰竭(13例,76.5%)。
以ILD起病的皮肌炎临床常见,是皮肌炎住院患者主要死亡原因。其主要临床病理类型是机化性肺炎和非特异性间质性肺炎。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
间质性肺疾病(ILD)是炎性肌病的常见而严重并发症,尤其在皮肌炎患者更为显著。皮肌炎患者并发ILD的发生率最高可达65%[1],病死率可高达40%[2]。相对来说,ILD的发生后于皮肌炎比较容易诊断,而对于ILD先于皮肌炎发生或与皮肌炎同时发生,即以ILD作为皮肌炎首发而突出表现者,则比较难于诊治。ILD先于炎性肌病发生者约占15%~20%,而ILD与炎性肌病同时发生者约占45%~65%,两者相加可达60%~80%[3,4,5]。关于以ILD起病的皮肌炎的临床特点国内外文献罕有涉及。本研究是对中国人群以ILD起病的皮肌炎患者的一项大样本回顾性研究。
北京协和医院1999年1月至2013年1月皮肌炎住院患者1 230例,其中合并ILD共210例。收集资料完整者184例,平均年龄(48±12)岁,病程中位数5个月(1~240个月),男∶女=63∶121,平均住院日(28±16)d。
皮肌炎的诊断需符合Bohan and Peter标准[6],包括:(1)对称性近端肌无力;(2)血清肌酶升高;(3)肌电图提示肌源性损害;(4)肌活体组织检查证实肌炎改变;(5)皮肌炎特征性皮疹。除外合并其他结缔组织病或者肌病。临床无肌病的皮肌炎(CADM)诊断符合Sontheimer标准[7],即具有典型皮肌炎皮疹,但是无肌病或者轻肌病表现。抗合成酶抗体综合征指炎性肌病患者同时伴抗合成酶抗体、发热、关节炎、雷诺现象、技工手和ILD[8]。抗Jo–1抗体是抗合成酶抗体中最常见的抗体(60%~80%)[8]。ILD的诊断有赖于高分辨CT(HRCT)检查发现符合ILD的表现(网状结节,磨玻璃或斑片,实变,不规则线状影,蜂窝或支气管扩张牵拉),并需除外下列疾病:(1)药物毒性作用;(2)肺结核;(3)结节病或肺部肿瘤;(4)长期吸烟或有其他环境暴露因素;(5)慢性支气管炎、肺气肿、肺源性心脏病、支气管哮喘、支气管扩张等呼吸系统疾病。
对本组皮肌炎合并ILD患者,收集并分析如下项目和指标:(1)临床特点,至少由一位风湿免疫科医生进行问诊查体;(2)血清学检查:抗核抗体、抗Jo–1及其他自身抗体,血清肌酶谱、血红细胞沉降率、免疫球蛋白IgG等;(3)影像学检查,至少由两位放射科医生读片;(4)血气分析及肺功能检查,肺功能检查主要记录用力肺活量和一氧化碳弥散率,结果以与预计值百分比表示,用力肺活量占预计值百分比< 80%为异常,一氧化碳弥散率占预计值百分比< 70%为异常;(5)肺活体组织病理学检查,经支气管镜、经皮肺穿刺或经外科手术获得肺组织;(6)治疗及转归。将本组皮肌炎合并ILD患者分为ILD起病组和非ILD起病组。ILD起病组包括ILD发病先于皮肌炎或者ILD与皮肌炎同时发病(ILD与皮肌炎伴随发生在1个月之内)[3]。非ILD起病组指ILD发病后于皮肌炎(ILD发生在皮肌炎发病后1个月以上)。比较两组患者上述临床特点、实验室检查以及治疗和转归之间的差别。
采用SPSS软件。计量资料比较用独立样本t检验(方差齐者用t检验,方差不齐者用校正t检验,非正态分布者采用秩和检验);计数资料比较用χ2检验(如期望频数<5,则用Fisher精确概率计算),以P<0.05为差异有统计学意义。
ILD在皮肌炎住院患者中的发生率为17%(210/1 230)。将入组的共184例皮肌炎合并ILD患者,分为ILD起病组和非ILD起病组。ILD起病组共88例(47.8%),男∶女= 32∶56,平均年龄(50±12)岁,病程中位数4个月(1~144个月)。非ILD起病组共96例(52.2%),男∶女= 27∶69,年龄(47±12)岁,病程中位数6个月(1~240个月)。两组患者年龄、性别比例及病程差异无统计学意义(P> 0.05)。
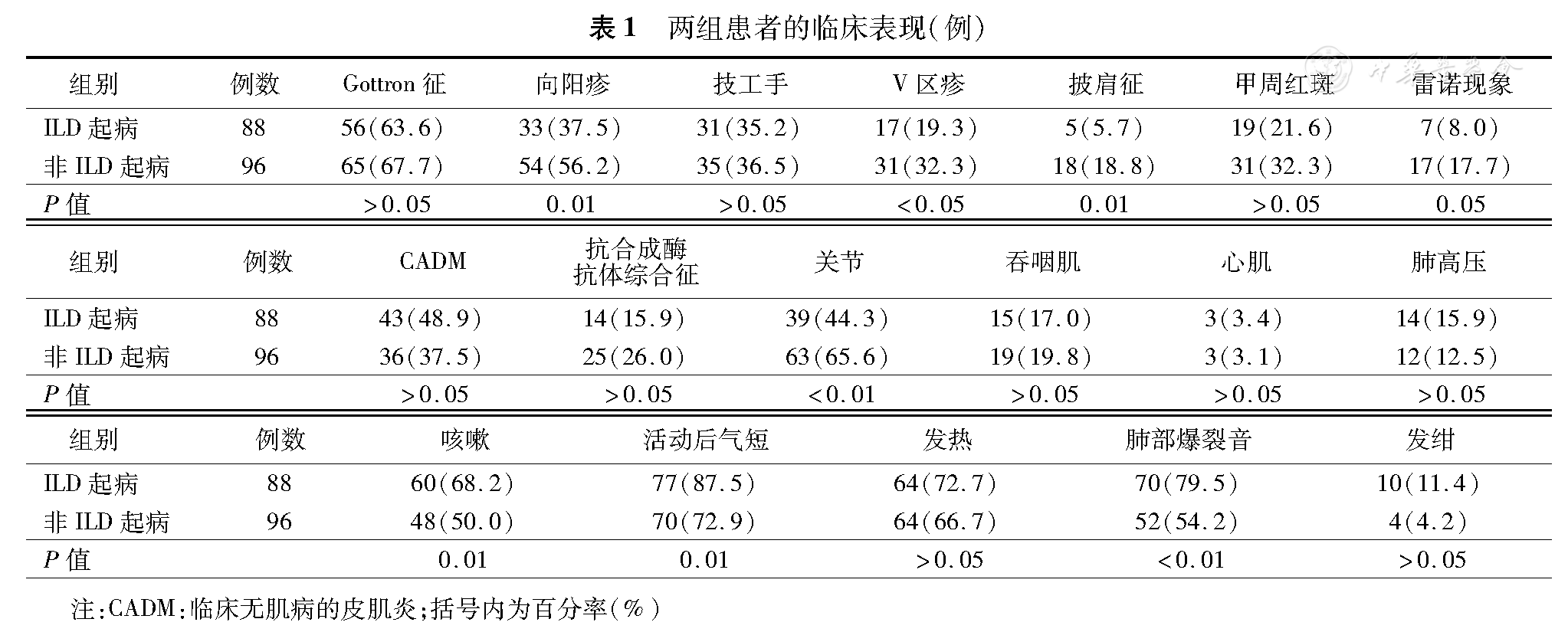
两组患者的临床表现(例)
两组患者的临床表现(例)
| 组别 | 例数 | Gottron征 | 向阳疹 | 技工手 | V区疹 | 披肩征 | 甲周红斑 | 雷诺现象 | CADM | 抗合成酶抗体综合征 | 关节 | 吞咽肌 | 心肌 | 肺高压 | 咳嗽 | 活动后气短 | 发热 | 肺部爆裂音 | 发绀 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ILD起病 | 88 | 56(63.6) | 33(37.5) | 31(35.2) | 17(19.3) | 5(5.7) | 19(21.6) | 7(8.0) | 43(48.9) | 14(15.9) | 39(44.3) | 15(17.0) | 3(3.4) | 14(15.9) | 60(68.2) | 77(87.5) | 64(72.7) | 70(79.5) | 10(11.4) |
| 非ILD起病 | 96 | 65(67.7) | 54(56.2) | 35(36.5) | 31(32.3) | 18(18.8) | 31(32.3) | 17(17.7) | 36(37.5) | 25(26.0) | 63(65.6) | 19(19.8) | 3(3.1) | 12(12.5) | 48(50.0) | 70(72.9) | 64(66.7) | 52(54.2) | 4(4.2) |
| P值 | >0.05 | 0.01 | >0.05 | <0.05 | 0.01 | >0.05 | 0.05 | >0.05 | >0.05 | <0.01 | >0.05 | >0.05 | >0.05 | 0.01 | 0.01 | >0.05 | <0.01 | >0.05 |
注:CADM:临床无肌病的皮肌炎;括号内为百分率(%)
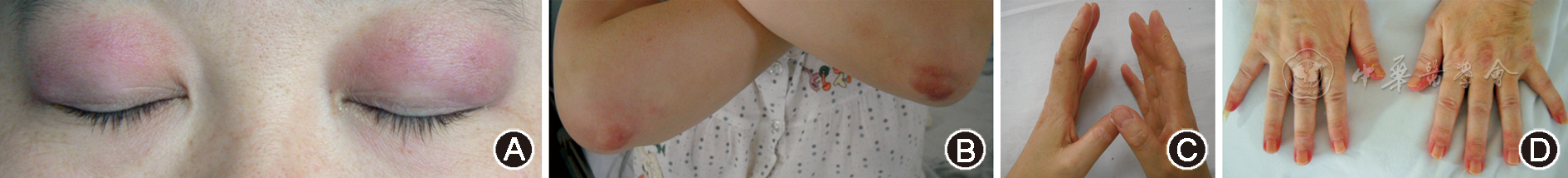
ILD起病组皮肌炎发生距离ILD发生的中位数时间为1个月(0个月~10年)。其中ILD与皮肌炎同时发生者共42例(22.8%),ILD先于皮肌炎发生者共46例(25.0%),平均先于皮肌炎(11±3)个月发生。ILD起病组包括CADM共43例(48.9%),抗合成酶抗体综合征14例(15.9%),与非ILD起病组相比差异无统计学意义(P> 0.05)。ILD起病组主要呼吸系统症状表现为活动后气短(77例,87.5%)、发热(64例,72.7%)、咳嗽(60例,68.2%)以及肺部爆裂音(70例,79.5%)。仅有2例患者无任何呼吸系统症状。其中,ILD起病组患者活动后气短、咳嗽和肺部爆裂音的发生率明显高于非ILD起病组(P< 0.05)。ILD起病组的皮肌炎患者皮疹(图1)包括向阳疹、前胸V区皮疹和披肩征的发生率显著低于非ILD起病组(P< 0.05),而雷诺现象发生率虽然与非ILD起病组相比差异无统计学意义(P= 0.05),但是却低于非ILD起病组(8%比17.7%)。两组之间Gottron征、技工手和甲周红斑发生率差异无统计学意义(P> 0.05)。肺外器官受累方面,ILD起病组的关节受累发生率显著低于非ILD起病组(P< 0.05),而其余器官受累如吞咽肌、心脏等发生率差异无统计学意义(P> 0.05)。
ILD起病组与非ILD起病组相比,血清肌酸激酶和乳酸脱氢酶平均水平更低,C反应蛋白水平更高(P< 0.05)。ILD起病组血红蛋白平均水平较非ILD起病组高(P< 0.05)。此外,ILD起病组血清抗核抗体和抗SSA阳性率显著低于非ILD起病组(P< 0.05),而两组抗Jo–1抗体阳性率差异无统计学意义(P>0.05),表2。
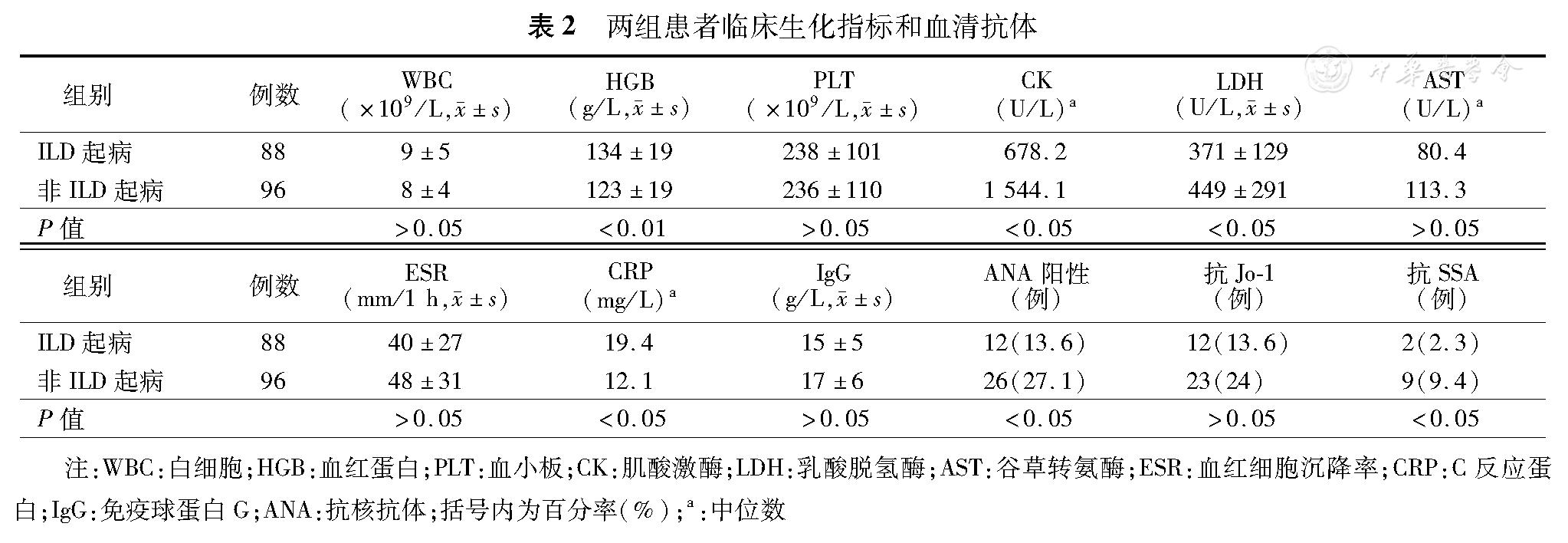
两组患者临床生化指标和血清抗体
两组患者临床生化指标和血清抗体
| 组别 | 例数 | WBC(×109/L, ±s) ±s) | HGB(g/L, ±s) ±s) | PLT(×109 /L, ±s) ±s) | CK(U/L)a | LDH(U/L, ±s) ±s) | AST(U/L)a | ESR(mm/1 h, ±s) ±s) | CRP(mg/L)a | IgG(g/L, ±s) ±s) | ANA阳性(例) | 抗Jo–1(例) | 抗SSA(例) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ILD起病 | 88 | 9±5 | 134±19 | 238±101 | 678.2 | 371±129 | 80.4 | 40±27 | 19.4 | 15±5 | 12(13.6) | 12(13.6) | 2(2.3) |
| 非ILD起病 | 96 | 8±4 | 123±19 | 236±110 | 1 544.1 | 449±291 | 113.3 | 48±31 | 12.1 | 17±6 | 26(27.1) | 23(24) | 9(9.4) |
| P值 | >0.05 | <0.01 | >0.05 | <0.05 | <0.05 | >0.05 | >0.05 | <0.05 | >0.05 | <0.05 | >0.05 | <0.05 |
注:WBC:白细胞;HGB:血红蛋白;PLT:血小板;CK:肌酸激酶;LDH:乳酸脱氢酶;AST:谷草转氨酶;ESR:血红细胞沉降率;CRP:C反应蛋白;IgG:免疫球蛋白G;ANA:抗核抗体;括号内为百分率(%);a:中位数
ILD起病组在HRCT上的主要表现包括:斑片影(63.6%)、磨玻璃影(51.1%)、网格(46.6%)、索条(39.8%)、实变(19.3%),其他少见表现还包括蜂窝影(10.2%)、纵隔气肿(5.7%)、肺大泡(4.5%)和结节(2.3%)等。
ILD起病组中81例进行血气分析检查,动脉氧分压平均(65±15) mm Hg,非ILD起病组(68±18) mmHg(1 mmHg=0.133 kPa),两组比较差异无统计学意义(P< 0.05)。ILD起病组共51例行肺功能检查,其中弥散功能障碍44例(86.3%),限制性通气功能障碍者43例(84.3%)。ILD起病组平均用力肺活量和一氧化碳弥散率占预计值的百分比分别为(60±12)%,(48±13)%,其中一氧化碳弥散率占预计值的百分比显著低于非ILD起病组(57±17)%,P<0.01。
ILD起病组中共31例(35.2%)患者行肺活体组织检查,检查手段包括经支气管镜肺组织活检(TBLB)24例,CT引导下经皮肺穿刺活检5例,经外科手术肺活检3例(包括2例胸腔镜手术、1例小开胸手术,其中1例先行TBLB,后行胸腔镜活检确诊)。其中17例(54.8%)提示肺泡间隔增宽、Ⅱ型肺泡上皮细胞增生和间质炎细胞浸润。7例(22.6%)符合机化性肺炎改变(其中4例通过TBLB,2例通过CT引导下经皮肺穿刺,1例通过外科开胸手术获得病理)。6例(19.4%)符合非特异性间质性肺炎改变(1例通过胸腔镜手术获得病理,5例通过TBLB)。另1例(3.2%)病理提示肺泡间隔增宽,间质大量淋巴细胞浸润及少量多核巨细胞,病变符合淋巴细胞性间质性肺炎。
ILD起病组全部88例患者均给予大剂量激素治疗(相当于泼尼松≥ 1 mg·kg–1·d–1)。其中21例(23.9%)因原发病皮肌炎病情需要,或肺部病变重且进展迅速而给予激素冲击治疗(甲泼尼龙1 g/d × 3 d)。21例激素冲击治疗的患者中共5例取得肺组织病理,其中2例病理证实为机化性肺炎。绝大多数患者(84例,95.5%)给予免疫抑制剂治疗,首选环磷酰胺,其他免疫抑制剂选择包括硫唑嘌呤、雷公藤、环孢素A等。ILD起病组17例皮肌炎患者死亡,病死率19.3%,与非ILD起病组病死率(19.8%)相比差异无统计学意义(P> 0.05)。本组经病理诊断为机化性肺炎的7例,好转6例,死亡1例;而经病理学诊断为非特异性间质性肺炎的6例和1例淋巴细胞性间质性肺炎均全部好转。经激素冲击治疗的21例好转15例。ILD起病组17例死亡患者的死亡原因包括ILD所致呼吸衰竭(13例,76.5%)以及合并感染(3例,17.6%),另有1例死亡原因为重度肺动脉高压猝死。
由于文献报道的研究人群年龄、人种、入选标准等因素的不同,ILD在炎性疾病的发生率也不尽相同,大约为35%~40%[3,4]。ILD可导致呼吸衰竭、肺高压及肺心病、继发肺部感染等,因此是皮肌炎患者的重要死亡原因[3,9,10]。炎性肌病并ILD患者5年病死率0~55%[2,3]。本研究发现ILD在皮肌炎住院患者中的发生率为17%,皮肌炎并ILD住院患者病死率高达近20%。因此,对皮肌炎患者并发ILD的早期识别和治疗尤为重要。但临床医生常可发现部分皮肌炎患者以ILD为首发而突出表现,这部分患者的临床诊治给风湿免疫科和呼吸科医生提出了挑战。
本研究发现,ILD起病先于皮肌炎者占皮肌炎并ILD住院患者的25%,而ILD与皮肌炎同时发生者占22.8%,两者相加共有近50%的皮肌炎并ILD住院患者是以肺部为首发起病的受累器官。这高度提示对于以ILD起病的患者,在问诊查体过程中应特别注意关注皮肤、骨骼肌表现,应进行肌酸激酶、皮肌炎相关的自身抗体检查以及其他必要实验室检查,以免漏诊。另一方面,由于25%的患者ILD可以先于皮肌炎出现,时间跨度1个月~10年(平均11个月),因此对于首诊时未能发现病因的ILD患者,应该在长期随诊过程中密切观察皮肤、骨骼肌症状,定期监测肌酸激酶变化,警惕出现皮肌炎。
本研究还发现,以ILD起病的皮肌炎患者呼吸系统症状突出,活动后气短、咳嗽和肺部爆裂音的发生率均显著高于非ILD起病的皮肌炎患者。因此,对于本组患者肺部病变的判断并不困难。此外,以ILD起病的皮肌炎患者向阳疹、前胸V区皮疹、披肩征和雷诺现象的发生率均低于非ILD起病组患者,所以,对于皮疹的观察随诊,以Gottron征、技工手、甲周红斑等更容易被发现。尽管两组CADM的发生率差异无统计学意义,但以ILD起病的皮肌炎患者肌酸激酶水平不如对照组升高明显,提示这部分患者骨骼肌症状可能较轻。
以ILD起病的皮肌炎患者血清抗核抗体和抗SSA抗体阳性率较低,但是抗Jo–1抗体的阳性率与对照组差异无统计学意义。因此,对于原因未明的ILD患者,应及时检测抗Jo–1抗体,可能使得部分患者能更早地被临床医生识别。Fischer等[11]提出"肺部优势结缔组织病(Lung–dominant connected tissue disease)"的概念,认为通过影像学或者病理诊断ILD的患者,如果除外其他病因,但又无任何结缔组织病肺外器官受累表现且不符合现有的任何结缔组织病的分类诊断标准时,必须进行血清自身抗体检查,如果存在高滴度抗核抗体阳性或其他结缔组织病特异性抗体,则可诊断肺部优势结缔组织病。该诊断标准中所指的特异性抗体,其中包括抗合成酶抗体。本结果表明,这部分符合肺部优势结缔组织病患者如果抗Jo–1抗体阳性,经过长期随诊观察很可能最终出现皮肌炎表现。
ILD起病组患者的ILD以机化性肺炎和非特异性间质性肺炎为主要临床病理类型。这两种临床病理类型都属于对激素治疗反应较好者。经过积极激素和免疫抑制剂(首选环磷酰胺)治疗,约80%患者病情好转或稳定。死亡的主要原因是由于ILD病情进展所致呼吸衰竭。遗憾的是,本研究发现通过各种方式取得肺组织的患者仅占1/3。临床上所采取的各种活体组织检查方式中,TBLB的确诊率不足50%,而外科手术病理学诊断的确诊率则能达100%。提示,应尽可能获得有效的肺组织活检以尽快明确病理类型,进而采取针对性有效的治疗方案,对于改善预后非常重要。

























