
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者女,30岁,因"口唇四肢不自主运动2年,加重3个月余"入院。患者2年前无明显诱因下出现口唇及四肢不自主运动,清醒时明显,入睡后消失;3个月前,患者症状加重,咬伤下唇,进食时将食物外推,并有口齿不清伴行走不稳,当地医院诊断考虑"肌张力障碍?强迫症?",予"氯硝安定"、"帕罗西汀"等治疗后改善不佳,遂至浙江大学医学院附属第二医院就诊。发病以来,患者体重减轻约10 kg。否认毒物接触史。父母为近亲结婚(表兄妹),有1兄,体健。体格检查:消瘦体型,角膜K-F环阴性,下唇变薄破溃,部分缺损,流涎(图1),可见口周不自主运动,口齿欠清,对答尚切题,双侧咽反射减弱,四肢肌张力减低,肌力5级,四肢不自主运动存在,指鼻试验稳准,双上肢反击征阳性,后拉试验阳性,行走时协调性差,双侧肱二头肌腱反射减弱,余腱反射未引出,双侧病理征阴性。
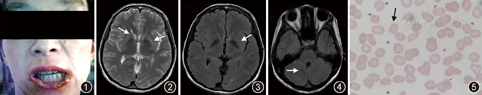
实验室检查:肌酸激酶:994 U/L(<145 U/L);肌酸激酶-MB:63 U/L(<24 U/L);天冬氨酸转氨酶:42 U/L(<34 U/L);乳酸脱氢酶:395 U/L(140~271 U/L);红细胞:3.50×1012/L [(3.68~5.13)×1012/L];血红蛋白:105 g/L(113~151 g/L);铜蓝蛋白、甲状腺功能、风湿免疫全套等均正常;其余化验无殊。头颅磁共振检查提示:两侧尾状核和壳核形态略缩小,考虑轻度萎缩;小脑齿状核及苍白球黑水相可见低信号影,考虑金属离子沉积。脑电图无殊。肌电图提示:(1)右尺运动神经传导速度减慢,右腓总神经运动传导远端潜伏期延长,运动传导诱发电位波幅降低;(2)右尺浅支、腓肠感觉神经传导速度减慢,右正中浅支、腓浅神经感觉传导诱发电位波幅降低;(3)右胫骨前肌检肌重收缩时运动单位数量减少。MMSE评分29分。
患者表现为突出的口周及四肢不自主运动,流涎,进食时将食物外推,肌酸激酶明显升高,头颅磁共振成像可见尾状核及壳核轻度萎缩(图2,图3,图4),综上,临床考虑神经棘红细胞增多症可能,予查外周血涂片可见棘红细胞,比例约55%(图5),故神经棘红细胞增多症诊断明确。予口服氟哌啶醇1 mg每天2次逐渐加量至2 mg每天2次,并补充维生素E等治疗后,复查肌酸激酶水平下降,症状较前改善,出院后随访12个月患者病情平稳。
神经棘红细胞增多症又称Levine-Critchle综合征[1],是一组罕见的以外周血中棘红细胞增多和进行性基底节退行性变为特征的神经系统遗传性疾病[2,3]。主要包括4种类型:舞蹈病-棘红细胞增多症、McLeod综合征、类亨廷顿病2型和泛酸激酶相关性神经退行性变。四者之间的鉴别要点见表1。
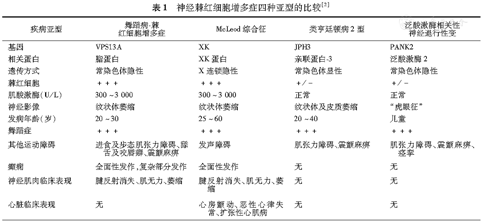
神经棘红细胞增多症四种亚型的比较[2]
神经棘红细胞增多症四种亚型的比较[2]
| 疾病亚型 | 舞蹈病-棘红细胞增多症 | McLeod综合征 | 类亨廷顿病2型 | 泛酸激酶相关性神经退行性变 |
|---|---|---|---|---|
| 基因 | VPS13A | XK | JPH3 | PANK2 |
| 相关蛋白 | 脂蛋白 | XK蛋白 | 亲联蛋白-3 | 泛酸激酶2 |
| 遗传方式 | 常染色体隐性 | X连锁隐性 | 常染色体显性 | 常染色体隐性 |
| 棘红细胞 | +++ | +++ | +/- | +/- |
| 肌酸激酶(U/L) | 300~3 000 | 300~3 000 | 正常 | 正常 |
| 神经影像 | 纹状体萎缩 | 纹状体萎缩 | 纹状体及皮质萎缩 | "虎眼征" |
| 发病年龄(岁) | 20~30 | 25~60 | 20~40 | 儿童 |
| 舞蹈症 | +++ | +++ | +++ | +++ |
| 其他运动障碍 | 进食及步态肌张力障碍、舔舌及咬唇癖、震颤麻痹 | 发声障碍 | 肌张力障碍、震颤麻痹 | 肌张力障碍、震颤麻痹、痉挛 |
| 癫痫 | 全面性发作,复杂部分发作 | 全面性发作 | 无 | 无 |
| 神经肌肉临床表现 | 腱反射消失、肌无力、萎缩 | 腱反射消失、肌无力、萎缩 | 无 | 无 |
| 心脏临床表现 | 无 | 心房颤动、恶性心律失常、扩张性心肌病 | 无 | 无 |
舞蹈病-棘红细胞增多症是本病最常见的类型,临床上主要表现为口面部不自主运动,咬唇癖,舞蹈样运动,癫痫发作,智能减退,认知功能障碍以及精神异常等;查体可有腱反射减低或消失,肌张力低下和肌肉萎缩[4]。血清肌酸激酶多增高,头颅磁共振可见尾状核、壳核萎缩,外周血涂片棘红细胞通常高于3%,但一些学者认为,棘红细胞阴性不能除外本病[5]。基因检测是确诊本病的"金标准"。但因技术限制,加上本病基因(VPS13A)信息量大,突变位点较多,检测上有一定难度[2]。因此,临床上提高对本病的认识很有必要。考虑到本例患者典型的临床特征、影像学表现及实验室检查,且其父母为近亲结婚,罹患遗传病的风险显著增加,舞蹈病-棘红细胞增多症诊断明确,暂未行基因检测。
本病主要与其他运动障碍性疾病如肌张力障碍、亨廷顿舞蹈病、肝豆状核变性等鉴别。在排除药物、毒物因素后,可进一步行血清铜蓝蛋白、尿铜水平测定,外周血涂片,肌电图,眼底镜检查,头颅磁共振成像,认知功能评定等帮助判断,必要时需行相应的基因检测。临床实践中,遇到口周不自主运动,肌张力障碍和舞蹈样动作的患者,需考虑到本病的可能,及时行外周血涂片检查,并注意家族遗传史的询问。
目前神经棘红细胞增多症的发病机制尚不清楚,可能因脂质代谢、铁代谢或线粒体功能异常所致[6]。本病治疗上尚无有效方案,以对症治疗为主,旨在提高患者生活质量,延长寿命。多巴胺受体阻滞剂(如氟哌啶醇)或多巴胺能耗竭剂(如丁苯那嗪)在改善运动过度及活动障碍上可能有一定疗效[2]。如出现癫痫发作可予抗癫痫药物,但卡马西平和拉莫三嗪可能加重不自主运动[7]。大剂量维生素E可改变红细胞膜的流动性,可能有助于病情的改善[8]。局部肉毒素注射可能改善局部症状。本病后期多有进食困难,常需留置胃管,必要时可考虑胃造瘘,还可出现营养不良,误吸,肺部感染等。本例患者接受氟哌啶醇及维生素E治疗后症状控制尚可,长期预后情况有待进一步随访。

























