
研究中国广东地区汉族产前人群中δ地贫球蛋白基因(HBD)的突变类型以及各类型突变频率,并统计分析各类型突变的血液学变化,以丰富人群δ-地贫基因突变图谱。
收集2012年1月至2015年5月在我院进行产前地中海贫血筛查的样本7 580例,筛选出符合条件的33例患者,男10例,女23例,年龄22~48岁。用XE 4000i血细胞分析仪进行血细胞分析,高效液相色谱法(HPLC)进行血红蛋白(Hb)、血红蛋白A2(HbA2)和血红蛋白F(HbF)定量检测。提取基因组DNA后进行PCR扩增、基因测序方法检测目标样本基因突变。
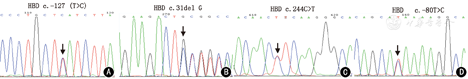
33例患者中检出21例存在δ-地贫突变,突变率约为0.277%(21/7 580)。检测出7种不同的突变类型。其中4种为已知的突变类型,包括12例基因型为-77(T>C)[HBD c.-127 (T>C)](57.14%),4例为-30 (T>C)[HBD c.-80 (T>C)](19.05%),1例为codon 10 (-G) (HBD c.31delG)(4.76%),1例为HBD c.244 C>T;另外新发现3种突变,1例为HBD c.22_24delGAG(4.76%),1例为HBD c.347 C>T(4.76%),1例为HBD c.349 C>G(4.76%)。
δ地贫基因突变类型多样,-77(T>C)是广东地区汉族产前人群最常见的δ球蛋白基因突变。本研究报道的3例新突变丰富了δ球蛋白基因的突变谱,应重视产前人群的δ球蛋白基因检测,对产前咨询和诊断有重要意义。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
地中海贫血是一种常染色体隐性遗传疾病。正常成人血红蛋白由血红蛋白A0(HbA0,α2β2,占比>96%)和血红蛋白A2(HbA2,α2δ2,占比2.5%~3.5%)及血红蛋白F(HbF,α2γ2,占比<1%)组成。发生在δ球蛋白基因(HBD)的突变或结构变异,将导致δ球蛋白合成减少,有的还伴随HbA2变异体[1]。通常β地中海贫血的患者由于β链合成减少而造成δ链合成相对增加,因此典型β地中海贫血的患者HbA2>3.5%。尽管δ蛋白基因的突变没有明显的临床表现[2,3],但当合并β地中海贫血时,由于δ链合成减少,使HbA2维持在正常水平甚至降低而掩盖了β地贫的存在,导致β地贫的漏诊[4,5]。华南地区人群β地中海贫血的突变携带率为2.54%[6],因此,检测HBD基因的突变是有必要的,尤其在产前人群中,夫妻一方有β地贫存在时。本研究通过对δ地中海贫血患者进行HBD基因检测,研究广东地区汉族产前人群中δ地贫的基因突变类型。
回顾性分析2012年1月至2015年5月在中山大学附属第一医院进行产前地中海贫血筛查的患者资料,从中选取符合以下条件的样本[7,8]:(1)血液样本高效液相色谱(HPLC)电泳结果HbA2<2.5%,伴或不伴有HbA2变异体的存在,或β地贫的HbA2低于预期;(2)HbA2<2.5%,且平均红细胞体积(MCV)≥27 fl和(或)平均红细胞血红蛋白量(MCH)≥81 pg;(3)排除缺铁性贫血或α地贫。所选样本入选标准为:(1)+(3)或(2)+(3)。共收集到符合条件患者33例,男10例,女23例,年龄22~48岁,均为广东地区汉族人群。所有受检对象采用乙二胺四乙酸二钾(EDTA-K2)真空抗凝管抽取静脉血2 ml。
血细胞分析仪(XE 4000i,日本Sysmex公司),高效液相色谱电泳仪(Variant Ⅱ,美国Bio-Rad公司),离心机(Eppendorf 5240,德国Eppendorf公司),全血基因组DNA提取试剂盒(Quickgene , Fujifilm,日本Kurabo公司),PCR扩增仪(ABI9700,美国ABI公司),凝胶成像分析系统(Vilber Louramt FUSION FX7,法国VILBER公司),水平电泳槽及电泳仪(美国Bio-rad公司)及恒温杂交仪(YN-H16,深圳亚能生物公司)。
红细胞血红蛋白量(Hb)、平均红细胞体积(MCV)、平均红细胞血红蛋白量(MCH)。
高效液相色谱电泳仪检测HbA0、HbA2、HbF水平。正常值:HbA0≥80%;HbA2:2.5%~3.5%;HbF: 0~3.5%。抽取外周静脉血2 ml进行血红蛋白电泳。收集HbA2<2.5%的样本。
δ地贫HBD基因检测的引物序列和PCR扩增参考文献[7]。
待测样品的PCR扩增产物电泳检测呈现单一清晰明亮条带,与标准带对照鉴定扩增产物片段大小和浓度是否符合测序要求,即可将待测样品直接送华大基因公司完成测序。目的片段序列通过chromas及clustal X软件与参考序列进行比对,找到突变位点后,进一步对异常结果做反向测序以验证结果的可靠性。
33份样本中,21份有δ地贫基因缺陷,阳性检出率为63.64%。
在来我院进行产前筛查的7 580例患者中,筛选出符合条件的患者33例,其中发现21例有δ地贫基因缺陷,δ地贫基因突变率约为0.277%(21/7 580)。
发现了7种缺陷类型,其中4种为已知的缺陷类型,3种为新发现的缺陷类型。最常见的突变类型为-77 (T>C),共12份样本检测出该类型,占57.15%(12/21);其次是-30 (T>C),占19.05%(4/21)。结果见表1。
南方汉族产前人群的δ球蛋白基因突变谱
南方汉族产前人群的δ球蛋白基因突变谱
| 基因型 | 例数 | 频率(%) | HbA2(%) | 位点 |
|---|---|---|---|---|
| -77 (T>C) | 12 | 57.15 | 1.6±0.2 | c.-127T>C |
| -30 (T>C) | 4 | 19.05 | 1.3±0.4 | c.-80 T>C |
| codon 10 (-G) | 1 | 4.76 | 1.6 | c.31delG |
| HbA2 Saint-Denis | 1 | 4.76 | 0.5 | c.244 C>T |
| codon8(p.Glu8del) | 1 | 4.76 | 1.9 | c.22_24delGAG |
| codon115(GCC>GTC) | 1 | 4.76 | 1.9 | c.347C>T |
| codon116(CGC>GGC) | 1 | 4.76 | 1.2 | c.349C>G |
注:HbA2:血红蛋白A2

21例δ地中海贫血患者的基因型和血液学分析结果见表2。
21例δ地中海贫血患者的基因型和血液学分析
21例δ地中海贫血患者的基因型和血液学分析
| 例序 | 性别 | 年龄(岁) | 基因型 | HbA2(%) | HbF(%) | Hb(g/L) | MCH(pg) | MCV(fl) |
|---|---|---|---|---|---|---|---|---|
| 1 | 女 | 35 | -77 (T>C) | 1.6 | 0.6 | 87 | 30.6 | 93.0 |
| 2 | 女 | 28 | -77 (T>C) | 1.6 | 2.1 | 143 | 29.1 | 83.1 |
| 3 | 女 | 26 | -77 (T>C) | 1.2 | 0.8 | 121 | 29.5 | 89.3 |
| 4 | 男 | 27 | -77 (T>C) | 1.7 | 1.2 | 157 | 29.3 | 84.7 |
| 5 | 男 | 36 | -77 (T>C) | 1.7 | 0.7 | 147 | 30.6 | 90.4 |
| 6 | 男 | 27 | -77 (T>C) | 1.6 | 0.6 | 137 | 29.5 | 84.9 |
| 7 | 女 | 25 | -77 (T>C) | 1.5 | 2.3 | 129 | 29.6 | 90.1 |
| 8 | 女 | 27 | -77 (T>C) | 1.8 | 4.3 | 119 | 31.5 | 89.4 |
| 9 | 女 | 35 | -77 (T>C) | 1.5 | 0.9 | 116 | 25.6 | 76.9 |
| 10 | 男 | 48 | -77 (T>C) | 2.1 | 5.5 | 58 | 34.3 | 105.9 |
| 11 | 女 | 27 | -77 (T>C) | 1.5 | 2.2 | 109 | 29.4 | 92.5 |
| 12 | 女 | 29 | -77 (T>C) | 1.4 | 1.6 | 127 | 27.7 | 83.0 |
| 13 | 女 | 26 | -30 (T>C) | 1.9 | 0.7 | 109 | 32.2 | 93.5 |
| 14 | 男 | 29 | -30 (T>C) | 0.8 | 0.3 | 159 | 30.6 | 87.7 |
| 15 | 女 | 28 | -30 (T>C) | 1.6 | 0.2 | 119 | 24.4 | 80.1 |
| 16 | 女 | 33 | -30 (T>C) | 0.9 | 0.7 | 121 | 31.5 | 89.3 |
| 17 | 女 | 30 | HbA2 Saint-Denis | 0.5 | 0.6 | 123 | 29.4 | 88.0 |
| 18 | 男 | 26 | codon10(-G) | 1.6 | 0.2 | 149 | 28.7 | 84.8 |
| 19 | 男 | 30 | codon116(CGC>GGC) | 1.5 | 0.2 | 166 | 30.2 | 85.2 |
| 20 | 男 | 37 | codon 115(GCC>GTC) | 1.9 | 0.3 | 160 | 31.4 | 90.6 |
| 21 | 女 | 25 | condon8(delGAG) | 1.9 | 0.4 | 119 | 30.7 | 90.7 |
注:HbA2:血红蛋白A2;HbF:血红蛋白F;Hb:血红蛋白;MCH:平均红细胞血红蛋白含量;MCV:平均红细胞体积
在成人阶段,δ珠蛋白的表达量约为β球蛋白的1/50,其与δ珠蛋白链构成HbA2。当出现δ珠蛋白基因突变或缺失时,δ珠蛋白链合成的质量或者数量下降,导致HbA2下降或者出现HbA2变异体。由于δ珠蛋白占人体总珠蛋白很少,所以δ地中海贫血没有明显临床表现。
迄今为止,有117种突变导致δ结构变异或导致δ地贫[9]。对于不同种群,δ地贫的基因缺陷类型以及各类型的发病率均有差异。Pavlou等[10]对希腊塞浦路斯人群δ地贫突变的研究统计出11种有义突变,其中最普遍的是HbA2-Yialousa,占所有缺陷类型的60.7%,其次是codon4C→T,占17.8%。Giambona等[8]对西西里人群δ地贫做了大量的研究,统计出12种缺陷类型,其中最普遍的也是HbA2-Yialousa,占81%,而且HbA2-Yialousa、HbA2 -NYU和IVS II nt 897 A→G共3种缺陷类型加起来占总缺陷的90%以上。Morgado等[11]对葡萄牙人群的研究发现6种δ球蛋白基因缺陷类型,其中HbA2-Yialousa也是最普遍的。对广东地区汉族产前人群的δ地贫研究显示,-77(T>C)[HBD -127(T>C)]突变是最常见的,这和其他国家和地区有显著差异,但与Liu等[7]和Xiong等[12]的结果一致。本研究中有12份样本携带此突变,占57.14%,这些样本的HbA2为(1.6±0.2)%。该突变位于δ球蛋白基因高度保守的核心序列区(GATA-1 motif),通过影响与GATA-1位点的结合使球蛋白基因表达下降[13]。其次是-30 (T>C)[HBD c.-80 (T>C)],占21例δ球蛋白基因缺陷的19.05%(4/21)。其HbA2为(1.3±0.4)%。该突变与δ球蛋白基因TATA盒有关,导致δ球蛋白基因转录减少,引起δ地中海贫血[14]。本研究发现1份样本为codon10(-G)(HBD c.31delG),其HbA2为1.2%,该移码突变使δ球蛋白在合成第94个氨基酸后合成提前终止,导致δ0地中海贫血。有1份样本为HbA2 Saint-Denis[delta 81(EF5) Leu>Phe,HBD c.244C>T][15],其HbA2为1.6%。
此外,本研究新发现了3种突变。其中1例为codon116(CGC>GGC)[δ116(G18)Arg(R) >Gly(G)](HBD c.349C>G),导致第116位密码子CGC转变为GGC,致使精氨酸转变为甘氨酸,该患者为男性,其HbA2为1.5%。1例为codon 115(GCC>GTC)[δ116(G17)Ala(A)>Val(V)],导致第115位密码子GCC转变为GTC,致使丙氨酸转变为缬氨酸,其HbA2为1.9%。1例为codon8(p.Glu8del)(HBD c.22_24delGAG),导致第八位密码子编码的谷氨酸丢失,其HbA2为1.9%。
本研究δ球蛋白基因缺陷阳性检出率为56.7%。其余12份样本,其HbA2<2.5%,且MCV、MCH正常,并除外缺铁性贫血和α地贫,但并未发现有δ球蛋白基因缺陷,这可能与引物设计有关,也可能是HPLC测量的HbA2不准确,或者δ球蛋白基因较大片段缺失,而测序未能测清。
通过对广东地区汉族产前人群中δ球蛋白基因的研究,发现其突变率约为0.277%,比Liu等[7]报道的0.4%略低,这可能与本研究样本量不够大和入选标准有关。这也意味着δ球蛋白基因缺陷在中国人群中是很罕见的。因此,对于HbA2水平降低但MCV正常的人群目前还不需要进行常规的δ球蛋白基因筛查。但对于那些MCV、MCH降低而HbA2水平正常或降低的患者在排除缺铁性贫血或者是δ地贫后,一方有此性状的夫妇,尤其另一方已经确诊为β地贫,需对患者做进一步的δ和β地贫基因分析,这样可以预防重型β地贫患儿的出生。

























