
分析尼曼匹克病C型(NPC)患者的临床表现和基因突变特点,观察疾病的治疗效果和预后。
收集2013年7月至2017年2月在首都医科大学附属北京天坛医院就诊的10例NPC患者,对其临床表现、常规化验检查和基因突变特点进行分析,并随访其治疗效果和预后。
10例NPC患者,男5例,女5例,就诊年龄42 d~14岁;包括2例新生儿型、4例早期婴儿型、2例晚期婴儿型和2例青少年型。所有患者均有明显的脾大,5例伴有肝大。头部磁共振示8例脑萎缩、2例胼胝体变细及2例脑白质区异常信号。10例患者共检出18种NPC1基因突变,其中11种为已知突变,另7种为新突变,包括3种错义突变:c.3683T>C (p.Met1128Thr)、c.1926G>C(p.Met642Iie)和c.3006C>G(p.Phe1002Leu);2种无义突变:c.1142G>A(p.Trp381Ter )和c.3229C>T(p.Arg1077Ter);1种微小缺失突变:c.1385-1386del;1种剪切位点突变:c.1757+5G>A。平均随访25(3~66)个月,丙咪嗪使5例患者的痴笑猝倒发作减轻,丙戊酸钠和卡马西平分别使2例和1例患者的癫痫发作程度减轻,随访结束时4例死亡,其余患者的病情均呈进行性加重。
NPC1基因突变谱具有高度异质性,中国人NPC1基因的突变热点可能与欧州国家不同。NPC患者的神经系统表现与其发病年龄有关。丙咪嗪对控制痴笑猝倒发作有效,卡马西平或丙戊酸钠可能在病程早期对控制癫痫发作有效。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
尼曼匹克病C型(NPC)是一种极少见的常染色体隐性遗传病,临床常出现广泛的内脏器官和神经系统受累。NPC在任意年龄均可发病,其病情进展速度与神经系统症状出现年龄有明显相关性,据此把NPC分为早期婴儿型(3个月~2岁)、晚期婴儿型(2~6岁)、青少年型(6~15岁)和成人型(>15岁),此外尚有以内脏受累为主的新生儿型(0~3个月)[1,2]。本研究对10例NPC患者的临床表现和基因突变特点进行分析,观察中国NPC患者的基因突变特点、临床特征、治疗效果及预后。
本研究为回顾性研究。病例资料来自2013年7月至2017年2月在首都医科大学附属北京天坛医院儿科就诊的患者,共有10例经基因检测确诊为NPC的患者,这些患者来自中国大陆不同地区,其中男5例,女5例,在北京天坛医院就诊年龄42 d~14岁。收集这10例患者的临床资料和实验室检查资料,包括血常规、血生化、胸片和腹部超声,9例行骨髓穿刺,8例行头部磁共振(MRI)检查。
采集患者及其父母静脉血各3~4 ml,采用高通量基因检测技术,对包括NPC1、NPC2、SMPD1和GBA等在内的系列遗传代谢病相关基因进行筛查。检测前签署知情同意书。采用FlexiGene DNA Kit试剂盒提取患者及家属静脉血全基因组DNA,用Nanodrop 2000超微量分光光度计测定DNA含量。用二代测序靶向捕获技术进行基因检测,用Agilent Sure Design在线工具设计针对目标基因外显子及侧翼序列(±10 bp)的靶向捕获探针,定制目标基因捕获试剂盒。采用目标序列富集试剂盒(Agilent SureSelect Target Enrichment System Kit)制备目标基因捕获文库(实验过程包括超声破碎DNA片段、文库构建、杂交捕获、捕获文库扩增和纯化),使用Agilent Bioanalyzer 2100仪器文库对DNA样本进行检测,应用NEXTSEQ500测序仪上机测序,测序的原始数据经RTA software、CASAVA software、BWA、Genome Analysis Toolkit生物信息软件分析,评估测序质量并生成单核苷酸变异报告,对候选变异采用Sanger法进行验证。
确诊后针对患者出现的临床表现采取个体化的对症治疗。出院后每隔3个月进行电话或邮件随访,每隔6~12个月门诊随访,了解病情进展并酌情调整用药;期间病情有明显变化,随时就诊或电话、邮件随访。
10例NPC患者中,2例在出生后2~3 d起病,平均就诊年龄1.5个月,主要表现为新生儿黄疸消退延迟、腹胀、纳差和体重增长缓慢,伴肌张力减低和轻度精神运动发育迟缓。8例以精神运动发育倒退或伴脾大为主诉就诊,平均就诊年龄为8.4(3.3 ~14.2)岁,这8例患者均有反复呼吸道感染史或多次肺炎史,神经系统症状的出现年龄在1.3 ~ 7.3岁,表现为进行性脑病、语言障碍及共济失调等,部分出现了典型的垂直性核上性眼肌麻痹(VGSP)、痴笑猝倒发作和肌张力障碍,伴或不伴有癫痫发作和睡眠障碍等(表1)。
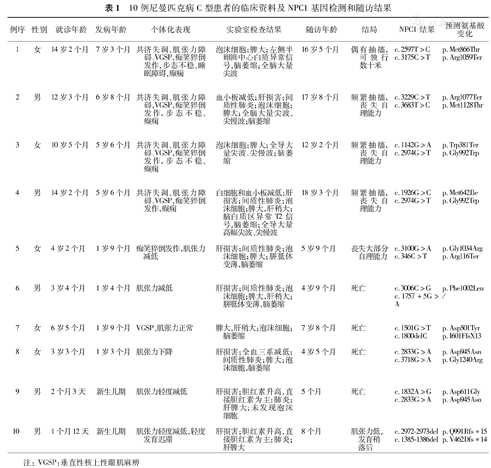
10例尼曼匹克病C型患者的临床资料及NPC1基因检测和随访结果
10例尼曼匹克病C型患者的临床资料及NPC1基因检测和随访结果
| 例序 | 性别 | 就诊年龄 | 发病年龄 | 个体化表现 | 实验室检查结果 | 随访年龄 | 结局 | NPC1结果 | 预测氨基酸变化 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 女 | 14岁2个月 | 7岁3个月 | 共济失调、肌张力障碍、VGSP,痴笑猝倒发作,步态不稳、睡眠障碍,癫痫 | 泡沫细胞;脾大;左侧半卵圆中心白质异常信号,脑萎缩;全脑大量尖波 | 16岁5个月 | 偶有抽搐,可独行数十米 | c.2597T>C | p.Met866Thr |
| c.3175C>T | p.Arg1059Ter | ||||||||
| 2 | 男 | 12岁3个月 | 6岁8个月 | 共济失调、肌张力障碍,VGSP,痴笑猝倒发作,步态不稳、癫痫 | 血小板减低;肝损害;间质性肺炎;泡沫细胞;脾大;全脑大量尖波、尖慢波;脑萎缩 | 17岁8个月 | 频繁抽搐,丧失自理能力 | c.3229C>T | p.Arg1077Ter |
| c.3683T>C | p.Met1128Thr | ||||||||
| 3 | 女 | 10岁5个月 | 5岁6个月 | 共济失调、肌张力障碍、VGSP,痴笑猝倒发作,步态不稳、癫痫 | 泡沫细胞;脾大;全导大量尖波、尖慢波;脑萎缩 | 12岁2个月 | 频繁抽搐,丧失自理能力 | c.1142G>A | p.Trp381Ter |
| c.2974G>T | p.Gly992Trp | ||||||||
| 4 | 男 | 14岁2个月 | 5岁6个月 | 共济失调、肌张力障碍、VGSP,痴笑猝倒发作,癫痫 | 白细胞和血小板减低;肝损害;间质性肺炎;泡沫细胞;脾大,肝稍大;脑白质区异常T2信号,脑萎缩;全导大量高幅尖波、尖慢波 | 18岁3个月 | 频繁抽搐,丧失自理能力 | c.1926G>C | p.Met642IIe |
| c.2974G>T | p.Gly992Trp | ||||||||
| 5 | 女 | 4岁2个月 | 1岁9个月 | 痴笑猝倒发作,肌张力减低 | 肝损害;间质性肺炎;泡沫细胞;脾大;胼胝体变薄,脑萎缩 | 5岁9个月 | 丧失大部分自理能力 | c.3100G>A | p.Gly1034Arg |
| c.346C>T | p.Arg116Ter | ||||||||
| 6 | 男 | 3岁4个月 | 1岁4个月 | 肌张力减低 | 肝损害;间质性肺炎;泡沫细胞;脾大,肝稍大;胼胝体变薄,脑萎缩 | 4岁9个月 | 死亡 | c.3006C>G | p.Phe1002Leu |
| c.1757+5G>A | / | ||||||||
| 7 | 女 | 6岁5个月 | 1岁9个月 | VGSP、肌张力正常 | 脾大,肝稍大;泡沫细胞;脑萎缩 | 7岁8个月 | 死亡 | c.1501G>T | p.Asp501Tyr |
| c.1800delC | p.I601FfsX13 | ||||||||
| 8 | 女 | 3岁3个月 | 1岁3个月 | 肌张力下降 | 肝损害;全血三系减低;间质性肺炎;脾大;泡沫细胞,脑萎缩 | 4岁5个月 | 死亡 | c.2833G>A | p.Asp945Asn |
| c.3718G>A | p.Gly1240Arg | ||||||||
| 9 | 男 | 2个月3天 | 新生儿期 | 肌张力轻度减低 | 肝损害;胆红素升高,直接胆红素为主;肺炎;肝脾大;未发现泡沫细胞 | 5个月 | 死亡 | c.1832A>G | p.Asp611Gly |
| c.2833G>A | p.Asp945Asn | ||||||||
| 10 | 男 | 1个月12天 | 新生儿期 | 肌张力轻度减低,轻度发育迟滞 | 肝损害;胆红素升高,直接胆红素为主;肺炎;肝脾大 | 8个月 | 肌张力低, 发育稍落后 | c.2972-2973del | p.Q991Rfs*15 |
| c.1385-1386del | p.V462Dfs*14 |
注:VGSP:垂直性核上性眼肌麻痹
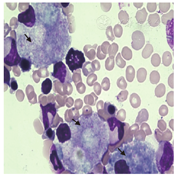
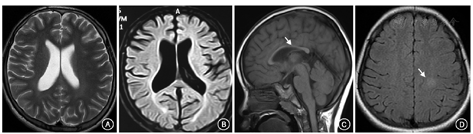
2例新生儿型NPC患者有明显的肝功损害、血胆红素升高、肺炎和肝脾大。8例以神经系统受累为主的患者中,3例出现血小板减少、白细胞减低或全血细胞减少,部分有肝功异常和间质性肺病,均有脾大,3例伴轻度肝大(表1),骨髓检查均发现有泡沫细胞(图1);在初次就诊或其后的随访中,头部MRI示进行性加重的脑萎缩(图2A,图2B),2例有胼胝体变薄(图2C),2例见脑白质区异常信号(图2D)。
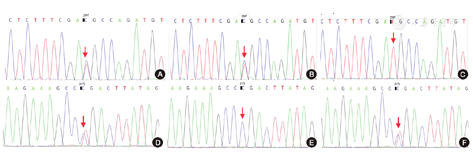
本组10例患者,在NPC1的两个等位基因中共发现18种形式的致病性突变,其中4种无义突变,3种缺失突变,1种内含子区剪切位点的突变,其余10种为错义突变。本研究发现了7种新突变:c.3683T>C (p.Met1128Thr)、c.1926G>C(p.Met642Iie)、c.3006C>G(p.Phe1002Leu)、c.1142G>A(p.Trp381Ter)、c.3229C>T(p.Arg1077Ter)、c.1385-1386del(p.V462Dfs*14)和c.1757+5G>A,既往未见公开报道,经查询1000Genomes和dbSNP数据库,发现这7种突变在人群中发生频率极低,不属于多态性变化,经Mutation Taster软件预测为致病性突变。
随访中发现,癫痫发作早期应用卡马西平和丙戊酸钠分别使2例和1例患者的癫痫发作程度减轻,随时间进展均又加重,更换或添加其他抗癫痫药物治疗,亦不能减轻癫痫发作,成为难治性癫痫。5例有痴笑猝倒发作的患者在服用丙咪嗪后,发作减轻,但目前的治疗均未能阻止病情的持续进展。随访25(3 ~ 66)个月,死亡4例,存活的6例患者病情均呈进行性加重,5例已丧失独立行走能力(其中1例为婴儿,尚未能独立行走),仅1例尚能独行数十米。
NPC是因NPC1或NPC2基因突变引起的常染色体隐性遗传病,据欧美国家对出生人口进行的大规模基因筛查估计,NPC出生人口发病率约在1/10万[3,4],其中95%是由NPC1突变所致,另有4%~5%与NPC2突变有关[1,3,5]。NPC1位于18q11-q12[6],有25个外显子[7],目前已发现的致病性突变超过300种(www.hgmd.cf.ac.uk)。在不同国家或地区,NPC1的突变热点不同,来自欧洲的报道中,最常见的是c.3182T>C(p.Ile1061Thr),占西欧地区报道总数的20%[8,9,10],其他常见突变位点还有c. 2324A>C (p.Gln775Pro)、c. 530G>A (p.Cys177Tyr)[10]和c. 3019C>G (p.Pro1007Ala)[9];报道的另一常见突变位点为c.2974G>T(p. Gly992Trp),主要来自加拿大[11,12]。本组10例患者共发现18种类型的NPC1突变,11种为已报道的致病性突变,例3和例4携带有c.2974G>T(p. Gly992Trp)的突变,与加拿大报道的突变热点相同,未发现欧洲国家报道的上述4个突变热点。本研究发现的7种新突变中,c.1142G>A和c.3229C>T为无义突变,c.1385-1386del为微缺失导致的移码突变,c.1757+5G>A为内含子区剪切位点的突变,预测上述突变对蛋白的空间结构和功能将产生重大影响。c.3683T>C、c.1926G>C和c.3006C>G为错义突变,预测的氨基酸变化为p.Met1128Thr、p.Met642IIe和p.Phe1002Leu,根据预测的氨基酸变化,对NPC1蛋白采用Polyphen-2软件进行评估,结果均为有害,有可能导致临床发病。
NPC的临床表现复杂多变,确立诊断需要有组织化学或分子遗传学结果的支持,但延误诊断的主要原因还是临床人员缺乏对NPC的基本认识[13,14]。延迟的新生儿黄疸/胆汁淤积、脾脏肿大、VGSP、痴笑猝倒发作和认知能力下降/痴呆等表现,是诊断NPC最强的提示因素[15,16]。
NPC在胎儿期即可表现为胎儿水肿或胎儿腹水。新生儿期发病多表现为广泛内脏受累的非特异性症状,常出现黄疸消退延迟、肝脾肿大、间质性肺病或伴有肌张力减低和精神运动发育迟滞[16]。本组中有2例在新生儿期发病,其临床表现与文献报道情况相符,基因检测发现这两例患者均携带有NPC1基因的复合杂合突变。随访过程中,例9的肝功损害呈进行性加重,黄疸持续未消退,胆汁酸持续增高,5月龄时死于肝功能衰竭。例10经对症支持治疗后,4月龄左右黄疸逐渐消退,6月龄时胆汁酸降至正常,仍有轻度的肝功损害和脾大,8月龄时神经发育评估显示有轻度精神运动发育迟滞。
早期婴儿型NPC患者,起病隐匿,发病初期主要表现为肝脾大或脾大,部分患者的肝脾大持续多年,直到出现神经系统症状而就诊时才被发现。晚发婴儿型或青少年型NPC患者的脾大可能在神经系统症状出现前数年已经存在[16],而肺部浸润导致的间质性肺病,也明显早于神经系统症状,部分患者在疾病早期还会出现肝大和肝功异常,这些儿童早期出现的症状,往往是疾病的首发症状[16],但各型NPC在内脏方面的表现缺乏特异性,而神经系统的表现差异较大[16]。本组4例早期婴儿型NPC患者的神经系统首先表现为精神运动发育倒退,而2例晚发婴儿型和2例青少年型NPC患者的共济失调、动作笨拙、饮水呛咳和构音障碍等小脑受累的症状出现较早,相对而言认知能力损害出现较晚。另外,早发婴儿型患者中有3例表现为肌张力减低,而在晚发婴儿型和青少年型患者却表现为明显的肌张力障碍。VGSP和痴笑猝倒发作在早期婴儿型患者中出现的频率均为1/4,也明显低于晚发婴儿型(2/2)和青少年型NPC患者(2/2),此外,本组病例中的癫痫发作也仅见于晚发婴儿型和青少年型患者。这些均提示NPC的神经系统表现具有高度异质性,其临床症候学与神经系统症状的出现年龄相关。

























