
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
很多病因可导致组织细胞损伤,包括糖尿病肾病、高血压肾病、狼疮肾炎、药物中毒和急性肾损伤迁延等,后继会发生组织细胞变性、坏死和炎症反应等。在损伤较小时,损伤灶周边正常细胞即启动再生修复,直至正常结构和功能完全恢复[1]。然而,一旦发生了较大面积的损伤或损伤次数过多,则会引起间质细胞外基质堆积填充组织区域的缺损,从而引起不可避免的组织纤维化[1]。纤维化疾病是复杂的病理过程,是以细胞外基质成分过度沉积、生成多于降解和组织重塑为特征的病理状态,可导致细胞的凋亡、增殖、转分化,最终多个器官功能发生衰竭。纤维化发生的主要器官包括心、肺、肝和肾。另外,有些器官组织在病理状态下,如皮肤瘢痕,炎性肠病,骨髓增生异常等也可发生纤维化[2]。在发达国家,大约一半的患者死于纤维化相关疾病,纤维化的发生和进程对于疾病死亡率起到了不可忽视的作用[3]。组织纤维化可发生于人体各种组织和器官中,引起结构的改变和功能衰竭,目前尚缺乏有效的治疗措施。近期研究发现表观遗传学修饰可调控纤维化的发生与发展[4,5,6]。因此,针对表观遗传学修饰调控机制,可能有助于开发抗纤维化的药物。
组蛋白甲基化是表观遗传学的重要组成部分[7],1984年,Richmond等[8]解析了核小体结构,即核小体的N端尾巴可以从核小体的球状核心上延伸出去,即组蛋白的尾巴。组蛋白的尾巴上存在多个赖氨酸或精氨酸位点,为甲基的结合提供了空间上的可能,从而介导基因的活化或关闭。目前组蛋白甲基化修饰在肿瘤调控中被发现参与了诸如转化生长因子(TGF)β、表皮生长因子受体(EGFR)等与纤维化共同的重要信号通路。然而,迄今为止针对这一修饰方式在纤维化疾病中的作用研究还为数不多。研究表明,组蛋白甲基化修饰参与了多器官成纤维细胞的活化和胶原物质沉积,而改变甲基化修饰活性可调节组织纤维化的发生和发展[5,9,10]。现就组蛋白甲基化修饰在组织纤维化的作用和机制及其潜在的靶向治疗意义阐述如下。
过去数十年间,组织纤维化发生发展的机制一直是国内外学者研究的热点,目前已知的主要病理机制包括间质成纤维细胞的激活导致α平滑肌肌动蛋白(α-SMA)和胞外基质(ECM)的过量产生,上皮-间充质转分化(EMT),氧应激以及炎症细胞的浸润与大量炎症因子的分泌[9]。ECM主要由活化的成纤维细胞产生,合成的产物主要有α-SMA、Ⅰ型胶原蛋白(collagen Ⅰ)和纤连蛋白(FN)[9]。EMT是一个上皮细胞失去极性逐步向间充质样细胞转化的过程[10]。在此期间,伴随着上皮细胞极性的丢失,E-钙黏蛋白表达下降,上皮细胞分裂周期停滞,转变为促纤维化表型[11]。转变为此表型的细胞产生大量促纤维化因子,如TGF-β、肿瘤坏死因子α(TNF-α)和血管内皮生长因子(VEGF)等[11]。与此同时,上皮细胞也产生大量炎症因子致使炎症细胞在组织浸润[12]。实验研究表明,炎症反应几乎伴随着整个纤维化的进程,并且随着炎症因子产生增多,内皮功能损伤加重,成纤维细胞的活化程度增高[13]。纤维细胞的活化与多条信号通路的激活密切相关[14,15],而组蛋白甲基化等表观遗传学修饰可参与致纤维化通路的激活,从而调控纤维化的发生与发展。
哺乳动物细胞内组蛋白甲基化修饰可引起转录抑制、转录激活、DNA损伤应答、转录延伸等。在组蛋白甲基化转移酶(HMT)的催化下,S-腺苷甲硫氨酸的甲基转移到组蛋白H3和H4上,与相应位点的赖氨酸或精氨酸残基进行共价结合,染色体构象发生改变[16],从而使基因表达呈多样性,调控疾病的发生发展。组蛋白甲基化由多种不同的组蛋白-赖氨酸甲基转移酶(HKMTs)与组蛋白-精氨酸甲基转移酶(PRMTs)催化。
早在20世纪60年代,科学家们就发现了组蛋白的赖氨酸可以被甲基化。目前发现的HKMTs中,除了HKMT4(Dot1)以外,都具有保守的SET结构域[17]。在已知的甲基化位点中,5个位点的研究最为深入,分别为组蛋白H3上的K4、K9、K27、K36和K79[17]。在一个赖氨酸位点上可以发生单甲基化、二甲基化和三甲基化修饰[18]。不同层次的甲基化修饰具有不同的功能,例如H3K4me3分布在基因的转录起始位点[19],而H3K4me1则分布在基因的增强子上[20]。在哺乳动物中,一共存在9种PRMTs,它们可以催化3种不同类型的精氨酸甲基化,其中包括单甲基化、不对称甲基化和对称的双甲基化[21]。近年有研究发现,HMT异常活化-组蛋白甲基化是引起组织纤维化的重要因素,为抗纤维化的治疗提供靶标[22]。表1列出了目前组蛋白甲基化在各组织纤维化中的作用及HMT抑制剂治疗纤维化疾病的可能机制。
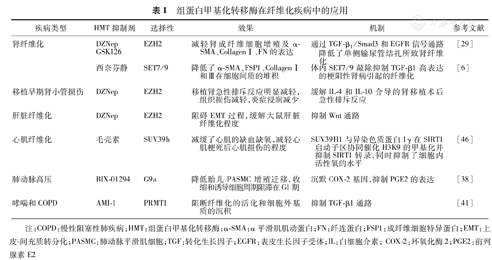
组蛋白甲基化转移酶在纤维化疾病中的应用
组蛋白甲基化转移酶在纤维化疾病中的应用
| 疾病类型 | HMT抑制剂 | 选择性 | 效果 | 机制 | 参考文献 |
|---|---|---|---|---|---|
| 肾纤维化 | |||||
| DZNep GSK126 | EZH2 | 减轻肾成纤维细胞增殖及α-SMA、CollagenⅠ、FN的表达 | 通过TGF-β1/Smad3和EGFR信号通路降低了单侧输尿管结扎所致肾纤维化 | [29] | |
| 西奈芬静 | SET7/9 | 降低了α-SMA、FSP1、CollagenⅠ和Ⅲ在细胞间质的堆积 | 体内SET7/9敲除抑制TGF-β1高表达的梗阻性肾病引起的纤维化 | [6] | |
| 移植早期肾小管损伤 | DZNep | EZH2 | 移植肾急性排斥反应明显减轻,组织损伤减轻,炎症浸润减少 | 缓解IL-4和IL-10介导的肾移植术后急性排斥反应 | |
| 肝脏纤维化 | DZNep | EZH2 | 阻碍EMT过程,缓解大鼠肝脏纤维化程度 | 抑制Wnt通路 | |
| 心肌纤维化 | 毛壳素 | SUV39h | 减缓了心肌的缺血缺氧,减轻心肌梗死后心肌损伤的程度 | SUV39H1与异染色质蛋白1γ在SIRT1启动子区协同催化H3K9的甲基化并抑制SIRT1转录,同时抑制了细胞内活性氧的水平 | [46] |
| 肺动脉高压 | BIX-01294 | G9a | 降低胎儿PASMC增殖迁移,收缩和诱导细胞周期阻滞在G1期 | 沉默COX-2基因,抑制PGE2的表达 | [38] |
| 哮喘和COPD | AMI-1 | PRMT1 | 阻断纤维化的活化和细胞外基质的沉积 | 抑制TGF-β1通路 | [41] |
注:COPD:慢性阻塞性肺疾病;HMT:组蛋白甲基化转移酶;α-SMA:α平滑肌肌动蛋白;FN:纤连蛋白;FSP1:成纤维细胞特异蛋白;EMT:上皮-间充质转分化;PASMC:肺动脉平滑肌细胞;TGF:转化生长因子;EGFR:表皮生长因子受体;IL:白细胞介素;COX-2:环氧化酶2;PGE2:前列腺素E2
糖尿病肾病是慢性肾脏病的主要病因,持续的高糖会引起肾小管上皮细胞坏死、转分化以及内皮细胞损伤,同时伴随着组蛋白甲基化转移酶SET7/9的过表达。SET7/9的甲基化位点为H3K9me2,一组来自于糖尿病患者的单核巨噬细胞数据显示,H3K9me2的改变与其免疫和炎症状态密切相关[23]。TNF-α可通过白细胞介素(IL)1A激活炎症反应[24]。进一步研究发现IL-1A在单核巨噬细胞中基础表达水平很低,而相比于单独TNF-α刺激,高糖与TNF-α共刺激下,细胞中的炎症反应会下降至少35%,究其原因可能为高糖诱导H3K9me2富集在IL-1A的启动子区并抑制转录,削弱了TNF-α介导的炎症反应。
同源序列2的增强子(EZH2),负责催化H3K27三价甲基化(H3K27me3)[25],而3-Deazaneplanocin A (DZNep)是针对EZH2活性的小分子抑制剂[26,27,28]。研究显示,EZH2的表达及其调控的H3K27me3与TGF-β刺激的大鼠肾成纤维细胞的活化程度呈正相关,并且在单侧输尿管结扎(UUO)的小鼠肾中也高表达;注射DZNep抑制EZH2活性和H3K27me3表达的同时,肾纤维化的程度也减轻。另外,DZNep处理也使肾脏细胞外信号调节激酶(ERK)、蛋白激酶B (AKT)、核因子κB(NF-κB)的磷酸化水平降低,表明EZH2-H3K27me3调控TGF-β和炎症通路激活肾纤维化[29]。
G9a催化了H3K9一价甲基化(H3K9me1)的发生。研究显示,经过UUO,7 d后小鼠肾脏G9a水平明显上调。在人类肾脏活检标本中,G9a免疫组化染色阳性区域的面积与H3K9me1及α-SMA和FN表达水平呈正相关。Klotho是成纤维细胞生长因子23(FGF-23)高亲和力受体的亚基,已有研究证明其可抑制TGF-β、Wnt及胰岛素样生长因子通路的激活。在UUO所致的肾纤维化模型中敲除G9a或使用bix01294,可以保留一部分Klotho并使肾纤维化程度降低,这是由于H3K9me1会富集在Klotho的启动子区域而抑制其转录[30]。以上证据提示G9a参与了慢性肾脏疾病,并且使用其抑制剂在纤维化的肾脏中具有治疗意义,这对开发抑制纤维化药物具有重要参考价值。
肝纤维化可由乙醇中毒、病毒和胆汁淤积等引起,其进展取决于肝星状细胞(HSC)转分化为肌成纤维细胞样表型的程度[2]。研究显示,乙醇直接诱导肝细胞弹性蛋白的表达,同时诱导HKMT-MLL1过表达使得H3K4甲基化而改变肝细胞的整个染色体结构,而H3K4甲基化可引起众多基因的开放。位点特异芯片分析证实在乙醇刺激下,HSC中MLL1/H3K4me3富集在原癌基因c-jun和成纤维细胞生长因子结合蛋白3(FGFBP3)基因处并激活[31]。提示乙醇可通过HSC的表观开放而引起纤维化。
富含胆碱的饮食可以调节组蛋白甲基化过程,因为胆碱对体内的腺苷及腺苷甲硫氨酸的浓度有重要影响,而S腺苷甲硫氨酸是细胞内甲基的主要供体[32]。缺乏胆碱的饮食喂养会导致大鼠肝组蛋白H3K9(H3K9me3)和H4K20的三价甲基化(H4K20me3)水平的降低,同时,伴随着HKMT-Suv39h1和Suv42h2水平的降低[32]。而H3K9me3在纤维化相关肝癌中起到了重要调控作用[33],外源性乙酰胆碱会引起HSC的增生并通过磷脂酰肌醇3-激酶(PI3K)和丝裂原活化蛋白激酶(MEK)信号通路激活α-SMA的表达增加[34]。由此可见,异常膳食也可导致机体发生表观遗传的改变,从而引起相应组织的纤维化。
肺纤维化主要因肺部炎症并引起随后的肺结构重塑,常见疾病有特发性肺纤维化、肺动脉高压、哮喘和阻塞性肺病[35]。目前尚缺乏有效的手段预防或逆转气道平滑肌增生肥大、黏膜化生、黏膜下和实质纤维化及持续炎症。
特发性肺纤维化是一种病因未明的致死性疾病,平均生存期限3~4年[36]。其病理特征为聚集和激活肺纤维细胞,激发过量胶原的沉积,导致肺脏结构的异常改变,肺功能进行性下降。特发性肺纤维化的发病与前列腺素E2(PGE2)和诱导型环氧化酶2(COX-2)缺乏有关,PGE2来自于COX-2。近期研究结果表明,G9a介导的H3K9位点和EZH2介导的H3K27位点的甲基化水平明显增加,并结合在COX-2的启动子区,导致肺纤维细胞COX-2的基因沉默;而G9a和EZH2的小干扰RNA(siRNA)可逆转其抑制性表观修饰,恢复PGE2和COX-2的产生[37]。这揭示了G9a和EZH2联合介导的组蛋白修饰在COX-2表观沉默中的作用,利于研究特发性肺纤维化的发病机制。
肺动脉平滑肌细胞(PASMC)过度增殖和凋亡会促进患者肺动脉高血压(PAH)的形成。在一项关于肺动脉平滑肌细胞增殖的研究中,Yang等[38]发现G9a参与了绵羊胎儿肺动脉平滑肌细胞增殖、迁移和收缩。使用BIX-01294降低胎儿PASMC增殖和诱导细胞周期阻滞在G1期。这一过程由p21介导,因为敲除p21基因可明显弱化BIX-01294抑制PASMC增殖的效应。同时BIX-01294治疗PASMC还抑制了血小板衍生生长因子(PDGF)诱导的细胞增殖、迁移和收缩。
尽管目前大多数的甲基化研究集中在组蛋白的赖氨酸位点,事实上,精氨酸位点的修饰也参与了纤维化过程的调节。在已发现的9种PRMTs中,PRMT1负责细胞内近85%的精氨酸甲基化[39]。PRMT1可引起组蛋白H4第3位精氨酸的二价不对称甲基化(H4R3me2a)[40]。有研究表明,PRMT1可激活TGF-β/COX-2和VEGF信号通路,在肺纤维化细胞中敲除PRMT1,COX-2和VEGF也随之下降。经气道吸入PRMT1的小分子抑制剂(AMI-1)治疗后,哮喘动物模型中肺脏PRMT1和COX-2表达水平均显著降低[41,42]。此外一项研究证实,PDGF-BB可通过增强ERK和STAT1的磷酸化刺激PRMT1表达,使用AMI-1抑制PRMT1活性下调了ERK通路激活的胶原纤维沉积和COX2的表达[43],更证实了精氨酸甲基化在肺纤维化中的表观调控作用。目前关于PRMTs调控纤维化疾病的研究较少,其是否能在其他组织纤维化中扮演调控角色有待探讨。
心脏纤维化会导致心脏肥厚硬化、心肌缺血缺氧和心律失常等。H3K79位点的甲基化与基因的开放有关,Delgado-Olguín等[44]发现EZH2缺乏则右心室明显纤维化且心内膜内陷。TGF-β3在野生型心肌细胞的核周低水平表达,而TGF-β3富集在EZH2基因敲除的心肌细胞外区域[45]。同源结构域转录因子Six1在骨骼肌细胞表达而非心肌细胞中。在心肌细胞中敲除EZH2,导致Six1基因过表达,心肌细胞会向骨骼肌细胞表型转分化并形成心肌肥厚。故EZH2可能通过抑制TGF-β3向胞外富集和沉默Six1表达来限制心肌肥厚和纤维化[44]。
HKMT-SUV39h可介导H3K9甲基化,SIRT1的表达上调可减轻心肌梗死,Yang等[46]发现缺血性或氧化应激致原代新生大鼠心室肌细胞中SUV39h的表达迅速上调,并且H3K9甲基化水平与SIRT1下调一致。相比于野生型小鼠,SUV39H基因敲除或使用SUV39h的抑制剂毛壳素的小鼠心肌梗死的程度明显减轻。SUV39h与异染色质蛋白1γ(HP1γ)在SIRT1启动子区协同催化H3K9的甲基化并抑制SIRT1转录,同时抑制了细胞内活性氧的水平。这些数据充分证明了SUV39h介导SIRT1的转录在心肌梗死中的作用。
近来的研究显示表观遗传学修饰可调控组织纤维化。组蛋白甲基化修饰可能促进纤维化相关基因的表达及促纤维分子的活化,进而诱使疾病的发生与发展。阻断组蛋白甲基化可以抑制组织纤维化并改善组织结构和功能,其机制可能与抑制TGF-β1/Smad3等通路、减少上皮细胞转分化及降低炎性反应等有关。揭示表观遗传修饰在组织纤维化发生发展中的作用和机制,将可能为治疗组织纤维化提供新靶点,为开发针对性的药物提供研发依据,对降低纤维化疾病患者死亡率,改善患者预后具有重要意义。

























