
探讨miR-106a-5p对血管内皮细胞的增殖、迁移功能的调控作用,及miR-106a-5p调节内皮细胞功能的可能作用靶点。
体外培养人脐静脉内皮细胞(HUVEC),通过逆转录实时荧光定量聚合酶链反应检测miR-106a-5p在HUVEC内的表达;并通过CCK-8实验、划痕实验检测miR-106a-5p对HUVEC增殖、迁移功能的影响;通过双荧光素酶实验及蛋白印记实验验证miR-106a-5p调节内皮细胞功能的可能靶点——信号转导与转录活化因子3(STAT3)。
miR-106a-5p可抑制HUVEC的增殖功能(F=13.62,P<0.01)。同时miR-106a-5p可抑制HUVEC的迁移功能:划痕12 h后,模拟物组的划痕闭合率下调(49.93/31.31)(χ2=8.240,P<0.05);划痕24h后,模拟物组的划痕闭合率下调(78.87/44.80)(χ2=10.50,P<0.01)。miR-106a-5p的直接靶基因为STAT3,并通过转录后作用调控STAT3的表达。
miR-106a-5p抑制HUVEC的增殖及迁移功能,其可能的作用靶基因为STAT3。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
动脉粥样硬化(AS)是一类由多病因导致的主要累及大中动脉的慢性炎症性疾病,而下肢动脉硬化闭塞症(ASO)是全身动脉粥样硬化性病变的重要组成部分,亦是血管外科的常见病,其可引起下肢长期慢性缺血,随着疾病进展,动脉管腔闭塞、侧枝循环无法代偿,最终肢体缺血、坏疽,严重者常需截肢,甚至有生命危险。
microRNAs是一类长度约21~23个核苷酸的内源性非编码小分子RNAs,通过与靶mRNA3′端非翻译区(3-untranslated region,3′-UTR)特异性结合,从而抑制该mRNA的翻译或直接降解该mRNA,以此调节蛋白质的表达[1,2]。作为AS病变重要参与者,内皮细胞(EC)、血管平滑肌细胞(VSMC)的功能均受到miRNAs的调控。本中心前期芯片结果提示,microRNA-106a-5p(miR-106a-5p)在ASO患者的血管壁表达降低[3]。本实验研究通过人脐静脉内皮细胞系探讨miR-106a-5p对内皮细胞功能的调控以及可能作用靶基因,以期为ASO的预防、治疗等带来了新的机遇。
人脐静脉内皮细胞(HUVEC)购自美国ATCC细胞库;人原代动脉平滑肌细胞分选自遗体捐赠的下肢股动脉。标本取材经中山大学附属第一医院医学伦理委员会批准。
胎牛血清(FBS)、DEME培养基、胰蛋白酶、磷酸盐缓冲液(PBS)购自美国Gibco公司;二甲基亚砜(DMSO)、结晶紫粉末购自美国Sigma公司;Trizol、4%多聚甲醛、LipofectamineTMRNA iMAX转染试剂、TEMED购自美国Invitrogen公司;miR-106a-5p引物、miRNA RT Mix试剂盒、miRNA实时聚合酶链反应(Real Time-PCR)试剂盒、STAT3引物、GAPDH引物购自大连Takara公司;miR-106a-5p模拟物(mimic)、抑制物(inhibitor)及阴性对照物均购自广州锐博生物科技有限公司;CCK-8试剂盒购自日本同仁化工;STAT3兔抗人一抗、p-STAT3兔抗人一抗、GAPDH兔抗人一抗、羊抗兔二抗、羊抗鼠二抗购自美国CST公司;Dual-Luciferase®Reporter Assay System购自美国Promega公司;ox-LDL购自广州奕源生物公司。
将细胞种于25cm2培养瓶中,采用10%FBS DMEM完全培养基,置于培养箱(37℃、5%CO2)中培养。每2~4天换液1次(根据细胞状态、密度及培养基颜色决定换液频率)。在普通倒置显微镜下观察细胞的生长状态,并根据实验目的,对细胞进行分组、种板,并予不同处理(如Ox-LDL刺激、转染、RNA提取、蛋白提取等)。
将细胞按0.5~1.5×106个/孔均匀种于6孔板中,置于培养箱中培养(37℃、5%CO2),每2~3天换液1次,待细胞密度达80%~95%时,更换培养基为DMEM,并根据既往文献报道,选取60μg/ml ox-LDL刺激内皮细胞[4,5]。
使用RNAiMAX转染试剂将miR-106a-5p mimic、抑制剂及模拟物阴性对照(NC)分别转染进生长状态良好、细胞密度达60%~70%的HUVEC,备后续实验。
制备miR-106a-5p引物,按照Trizol法提取细胞总RNA,使用miRNA RT Mix试剂盒及mRNA RT Master Mix进行逆转录,同时miRNA Real Time-PCR试剂盒进行聚合酶链反应。
将生长状态良好的HUVEC种于96孔板(5 000~6 000个/孔),培养箱(37℃、5%CO2)中培养约24 h后,进行转染(miR-106a-5p mimic及NC);8~12 h后,更换1% FBS DMEM饥饿24 h;更换10%FBS DMEM完全培养基继续培养24 h;弃去培养基,每孔加入100 μl细胞增殖毒性检测试剂盒(CCK-8)溶液(CCK-8试剂:10%FBS DMEM=1∶9);以锡纸包裹培养板(避光),继续培养,1~4 h后使用酶标仪检测各孔的吸光度(A)值(酶标仪调至450nm波长)。
将处于对数生长期的HUVEC种于6孔板(0.5~1.5×106个/孔),培养箱(37℃、5%CO2)中培养约24 h后,进行转染(miR-106a-5p mimic及negative control);转染8~12 h后,予10%FBS DMEM培养;至细胞密度达95%~100%时,更换1%FBS DMEM饥饿24 h;以200 μl无菌枪头,在每孔纵向划痕;弃去旧培养基,更换10%FBS DMEM完全培养基,在0、6、12、24h时于倒置荧光显微镜(白光)下观察拍照倒置。
以蛋白裂解液提取细胞总蛋白,检测各样本蛋白浓度并配平;使用8%、10%分离胶及5%积层胶进行蛋白印记实验,转膜并孵育一抗、二抗后进行曝光。
miR-106a-5p的碱基序列为AAAAGUGCUUACAGUGCAGGUAG,通过TargetScan、miRBase等数据库预测发现,STAT3具有3个能够与miR-106a-5p碱基互补(UUUCACG)的位点(158~164、448~454、623~629),提示STAT3可能是miR-106a-5p的靶基因。构建含有目的DNA片段(STAT3)的野生载体及突变载体(GCACTTT突变为CGTGAAA),其中突变载体含有上述3个位点的突变;位点一(252~258)、位点二(542~548)、位点三(717~723)。双荧光素酶报告体系(Dual-Luciferase®Reporter Assay System)验证miR-106a-5p与STAT3的靶基因关系。
结果以SPSS 19.0统计分析软件进行分析。分析前检验数据的方差齐性,方差齐时使用t检验或方差分析;方差不齐时使用校正t检验;均数间的两两比较,应用Bonferroni法检验。P<0.05为差异有统计学意义。
miR-106a-5p在HUVEC中的表达较其在血管平滑肌细胞中的表达丰富(4.41/1.00)(图1),差异有统计学意义(t=4.964,P<0.05)。
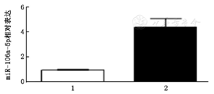
1:血管平滑肌细胞;2:人脐静脉内皮细胞
ox-LDL刺激HUVEC建立体外内皮损伤模型,结果显示,经过ox-LDL刺激后,HUVEC中miR-106a-5p的表达量下降(F=54.34,P<0.001);经过ox-LDL刺激12 h后,miR-106a-5p的表达下调1.85倍(1.00/0.54)(t=7.820,P<0.05),刺激24 h后,miR-106a-5p的表达下调2.39倍(1.00/0.42)(t=9.881,P<0.05),但是ox-LDL刺激HUVEC 12 h与24 h后,miR-106a-5p的表达差异无统计学意义。
miR-106a-5p可抑制HUVEC的增殖功能(F=13.62,P<0.01),但抑制作用并不显著,且与miR-106a-5p的模拟物(mimic)浓度相关。与阴性对照组相比,转染不同浓度(50、100 nmol/L)的miR-106a-5p模拟物后,HUVEC增殖功能受到抑制(1.309/1.126、1.309/1.125)差异均具统计学意义(t=3.446、3.460,P<0.05);但转染20 nmol/L的模拟物后,HUVEC的增殖功能则无明显改变(1.309/1.240),差异无统计学意义。
miR-106a-5p可抑制HUVEC的迁移功能(图2、图3)。划痕12、24 h后,模拟物组的划痕闭合率较空白对照组分别下调(49.93/31.31、78.87/44.80),差异均有统计学意义(χ2=8.240,χ2=10.50,P<0.05)。


1:模拟物阴性对照组1;2:miR-106a-5p模拟物;3:模拟物阴性对照组2;4:miR-106a-5p抑制剂
miR-106a-5p(previous ID:miR-106a)的碱基序列为AAAAGUGC UUACAGUGCAGGUAG,通过TargetScan、miRBase等数据库预测发现,STAT3具有3个能够与miR-106a-5p碱基互补(UUUCACG)的位点(158~164、448~454、623~629),提示STAT3可能是miR-106a-5p的靶基因。构建含有目的DNA片段(STAT3)的野生载体及突变载体(GCACTTT突变为CGTGAAA),其中突变载体含有上述3个位点的突变:将靶序列位点一:GCACTTT突变为CGTGAAA(252~258)、位点二:GCACTTT突变为CGTGAAA(542~548)、位点三:GCACTTT突变为CGTGAAA(717~723)。共转染miR-106a-5p模拟物和野生型STAT3 3′UTR质粒后(WT-STAT3 3′UTR),HEK 293细胞的荧光素酶活性降低(3.148/1.782),差异有统计学意义(F=7.837,P<0.05);共转染miR-106a-5p mimic和突变型STAT3 3′UTR质粒后(MUT-STAT3 3′UTR),HEK 293细胞的荧光素酶活性无明显改变(F=1.980,P>0.05),因此miR-106a-5p的靶基因可能为STAT3。共转染miR-106a-5p模拟物和野生型STAT3 3′UTR质粒后HEK293细胞的荧光素酶活性降低。
与阴性对照组相比,50 nmol/L及100 nmol/L的miR-106a-5p模拟物均对STAT3的mRNA表达无明显影响(t=0.0835/0.2038,P>0.05)(图4)。
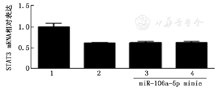
1:空白对照组;2:模拟物阴性对照组:3∶50 nmol/L的miR-106a-5p模拟物;4::100 nmol/L的miR-106a-5p模拟物
miR-106a-5p模拟物(50、100 nmol/L)抑制STAT3的蛋白表达(F=11.16,P<0.01),同时STAT3磷酸化形式p-STAT3的蛋白表达亦下调(F=6.305,P<0.05)(图5)。而miR-106a-5p抑制物可上调STAT3的蛋白表达(F=19.70,P<0.01)(图6)。
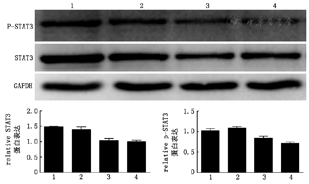
1:空白对照组;2:模拟物阴性对照组:3∶50nmol/L的模拟物;4:100nmol/L的模拟物
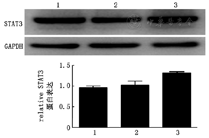
1:空白对照组;2:模拟物阴性对照组:3:miR-106a-5p抑制剂(100 nmol/L)
构建STAT3的干扰RNA(siRNA),通过划痕实验显示,在siRNA沉默STAT3后,HUVEC的迁移功能受到抑制(图7)。划痕12 h后,siRNA组的划痕闭合率较空白对照组下调(46.53/22.71),差异有统计学意义(χ2=9.50,P<0.01);划痕24 h后,siRNA组的划痕闭合率较空白对照组下调(86.54/48.18),差异有统计学意义(χ2=10.82,P<0.01)。

在AS的发生、发展过程中,EC发挥了重要作用,当EC在多种因素作用下发生功能失调,被激活的EC表达多种白细胞黏附因子,致单核细胞/巨噬细胞募集于血管内膜表面并迁移进血管壁,之后泡沫细胞形成、脂质条纹积累、VSMC迁移聚集、纤维帽形成[6,7,8]。同时EC的增殖、迁移与血管生成密切相关。而新生血管一方面可以增加缺血组织的血流灌注,另一方面亦在促进动脉粥样斑块增大、破裂等病理过程中扮演重要角色[9,10]。
STAT3是由STAT3基因编码的转录因子,属于STAT家族,STAT与其上游Janus kinase(JAK)构成JAK/STAT信号通路,影响其下游多种基因的表达,参与多种病理生理过程[11,12]。JAK/STAT通路和细胞炎症以及血管生成关系密切。已有研究证实,在动脉硬化组织中,JAK/STAT通路处于激活状态[13];干扰素(IFN)-γ和TLR4间通过STAT1的沟通形成了信号网络,从而将促AS的作用进一步放大[14]。
本中心前期数据芯片显示,miR-106a-5p在ASO者的血管壁表达降低[3],内皮细胞及平滑肌细胞均参与构成血管壁,由于捐赠标本获取有限,因此在体外实验中,选取人脐静脉内皮细胞系及原代培养的人平滑肌细胞进行qRT-PCR分析显示,miR-106a-5p在人脐静脉内皮细胞系中表达较为丰富,因此本研究优先选择在人脐静脉内皮细胞系中探讨miR-106a-5p的功能。通过体外实验证实miR-106a-5p抑制内皮细胞的增殖及迁移,可认为其作为血管生成抑制因子,通过在ASO患者的血管壁中表达下调发挥一定的血管保护作用。进一步的双荧光素酶报告,及针对靶基因STAT3的干扰实验,则显示miR-106a-5p可能通过靶基因STAT3调解内皮细胞的迁移等功能参与疾病的进展,这与既往研究相符[13,14]。但下肢动脉硬化闭塞症的疾病过程由多种细胞及介质参与,同时在疾病的不同阶段内皮细胞可能发挥不同的功能,因此miR-106a-5p在ASO患者中的作用尚需进一步的体内实验探索。
所有作者均声明不存在利益冲突

























