
评价注射用重组抗人表皮生长因子受体2(HER2)人源化单克隆抗体(赛普汀)联合长春瑞滨用于HER2阳性转移性乳腺癌的临床疗效和安全性。
受试者按2∶1比例随机至试验组和对照组,试验组接受赛普汀(首剂4 mg/kg,维持剂量每周2 mg/kg,静脉滴注)联合长春瑞滨(25 mg/m2,第1,8,15天/28天,静脉滴注)治疗;对照组接受长春瑞滨(25 mg/m2,第1,8,15天/28天,静脉滴注)化疗。主要研究终点为无进展生存期(PFS)。
2009年1月至2013年1月期间共纳入受试者315例(试验组212例,对照组103例)。试验组较对照组中位PFS显著延长,为39.1周比14.0周(HR=0.24;95%CI,0.16~0.36;P<0.000 1)。试验组客观缓解率(ORR)和疾病控制率(DCR)较对照组均显著提高,ORR为46.7%比18.45%(P<0.000 1),DCR为79.72%比45.63%(P<0.000 1)。中性粒细胞减少、白细胞减少和红细胞减少在两组发生率均较高,但组间差异无统计学意义。与赛普汀相关的不良反应最常见的为输注反应。共5例受试者治疗中心脏左室射血分数降低至低于50%,均可恢复,未出现严重心脏毒性。
赛普汀联合长春瑞滨具有显著疗效和良好安全性,是用于紫杉类治疗后的HER2阳性晚期乳腺癌的优选方案,为中国HER2阳性乳腺癌患者提供了更多靶向治疗机会。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
人表皮生长因子受体2(HER2)阳性乳腺癌约占乳腺癌整体的20%~30%,对于此亚型,HER2基因过表达可能是肿瘤发生发展的驱动因素。抗HER2靶向治疗于1998年第一个抗体药物曲妥珠单抗(赫赛汀)应用于临床,2002年开始在中国使用,直至2008年,对于大部分中国患者而言仍是不可及药物,临床实践中许多HER2阳性乳腺癌患者无法负担治疗费用而放弃抗HER2靶向治疗。本研究基于临床治疗现状和治疗需求进行试验设计,纳入HER2阳性复发转移性乳腺癌,未使用过抗HER2靶向治疗,但绝大多数受试者在复发转移后接受化疗治疗失败。
中国自主研发的注射用重组抗HER2人源化单克隆抗体(赛普汀)创新药,为非生物类似药,由三生国健药业(上海)股份有限公司(原中信国健药业公司)生产,于2004年7月2日经中国国家食品药品监督管理局批准进行临床研究(批件号2004L02352)。前期研究已证实赛普汀联合长春瑞滨治疗的显著有效性和良好安全性。本研究为临床多中心随机对照研究Ⅲ期临床试验,由解放军总医院第五医学中心(原军事医学科学院附属医院)为研究牵头单位,联合全国共26个研究中心参加,目的是评价赛普汀联合长春瑞滨用于紫杉类治疗后,但未经抗HER2治疗的HER2阳性转移性乳腺癌的临床疗效和安全性,同时给无法接受赫赛汀治疗的患者提供抗HER2靶向治疗机会。
本研究是一项随机对照、多中心、前瞻性Ⅲ期临床试验,筛选合格的受试者按2∶1比例随机分配至试验组和对照组。主要入选标准:(1)年龄18~70岁;(2)经组织病理学确诊的晚期乳腺癌患者;(3)HER2过度表达,经免疫组织化学法(IHC)检测为+++,或者荧光杂交法(FISH)检测为阳性;(4)按照实体瘤的疗效评价标准(RECIST),有明确可测量病灶;(5)既往使用细胞毒药物治疗不超过三线;(6)允许中枢神经系统转移患者入组,但需稳定。主要排除标准:(1)既往使用过同类品种的单克隆抗体,如曲妥珠单抗;使用过长春瑞滨;(2)心功能左室射血分数(LVEF)<50%或心脏疾病患者;(3)症状未控制的脑转移的患者;(4)4周内进行过放疗或化疗或参加过其他药物临床研究的患者。临床试验进行前研究方案经医院伦理委员会批准,所有患者在筛选前充分了解试验相关内容并签署知情同意书。
试验用药物为注射用重组抗HER2人源化单克隆抗体(赛普汀)(50 mg/支,三生国健上海药业公司,生产批号20081201,20090601)以及重酒石酸长春瑞滨注射液(10 mg/支,法国皮尔法伯制药公司,生产批号2AQ1083,2AQ1086,2AQ1087)。试验组接受赛普汀[(首剂4 mg/kg,维持剂量每周2 mg/kg,静脉滴注(静滴)],联合长春瑞滨(25 mg/m2,第1,8,15天/28天,静滴),治疗至疾病进展,如化疗毒性不能耐受,继续赛普汀单药治疗(首剂8 mg/kg,维持剂量每3周6 mg/kg,静滴),直至疾病进展。对照组接受单药长春瑞滨化疗(25 mg/m2,第1,8,15天/28天,静滴),直至出现疾病进展或化疗毒性不能耐受,序贯至赛普汀单药治疗(首剂8 mg/kg,维持剂量每3周6 mg/kg,静滴)直至疾病进展。
在试验期间,受试者定期接受生命体征、体格检查和相关实验室检查,每4周进行1次安全性评估,每8周进行1次疗效评估。试验组或对照组受试者进入赛普汀单药治疗后,则每6周进行1次安全性评估和疗效评估。采用RECIST 1.0进行疗效评价。主要研究终点为无进展生存期(PFS),定义为从受试者第1次用药到第1次记录的肿瘤进展或者任何原因导致死亡的时间。次要研究终点为客观缓解率(ORR),定义为最佳疗效完全缓解(CR)和部分缓解(PR)的受试者人数占总人数的百分比。疾病控制率(DCR),定义为最佳疗效CR+PR+疾病稳定(SD)的受试者人数占总人数的百分比。
安全性评价包括不良事件(AE),严重不良事件(SAE),心脏毒性,按照新的药物不良反应判定标准(NCI-CTC2.0)进行评估。AE:是指在使用药物过程中或用药后发生的不可预见的医疗状况或原有的医疗状况恶化,无论其是否与研究药物有关。SAE:是指发生在研究任何阶段,使用任何剂量的研究药物和(或)对照药物的AE,且其符合下列一个或者多个标准:(1)导致死亡;(2)危及生命;(3)导致住院或延长住院时间;(4)永久或显著丧失功能/残疾;(5)致畸或致癌。心脏毒性:从临床心脏相关症状,心电图异常,心脏LVEF下降三方面进行评估。
采用logrank检验比较两组中位数M[Q1,Q3]PFS的差异,如P<0.05表示试验组的中位生存时间好于对照组;并采用COX回归分析计算两组HR值的95%置信区间(CI),COX回归模型中考虑的因素包括组别及中心,如果HR的95%CI上限<1表明试验组的中位生存时间好于对照组。此外进行COX回归的敏感性分析,COX回归模型纳入的因素包括组别、是否内脏转移、DFS(≤2年与>2年)、激素受体状态[雌激素受体(ER)或孕激素受体(PR)阳性与ER和PR均阴性)以及年龄。采用χ2检验或Fisher精确概率法比较两组ORR的差异,并计算两组率差的95%CI,以相同的方法统计两组DCR的差异。统计方法均为双侧检验,校正水平α=0.05。
本研究于2009年1月开始,至2013年1月结束。共纳入26个研究中心,随机化入组受试者341例,其中试验组228例,对照组113例。进入全分析数据集(FAS)受试者315例,其中试验组212例,对照组103例。进入安全性数据集(SAS)受试者332例,其中试验组225例,对照组107例。受试者基线特征见表1。全分析数据集定义:是按照意向性分析(ITT)原则确定的数据集,包括所有随机化入组、至少使用过1次试验药的病例。安全性数据集定义:使用过试验药并至少有一次安全性评价记录的病例。
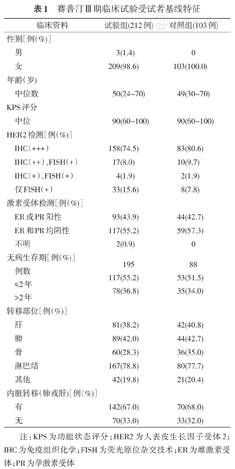
赛普汀Ⅲ期临床试验受试者基线特征
赛普汀Ⅲ期临床试验受试者基线特征
| 临床资料 | 试验组(212例) | 对照组(103例) | |
|---|---|---|---|
| 性别[例(%)] | |||
| 男 | 3(1.4) | 0 | |
| 女 | 209(98.6) | 103(100.0) | |
| 年龄(岁) | |||
| 中位数 | 50(24~70) | 49(30~70) | |
| KPS评分 | |||
| 中位 | 90(60~100) | 90(60~100) | |
| HER2检测[例(%)] | |||
| IHC(+++) | 158(74.5) | 83(80.6) | |
| IHC(++)、FISH(+) | 17(8.0) | 10(9.7) | |
| IHC(+)、FISH(+) | 4(1.9) | 2(1.9) | |
| 仅FISH(+) | 33(15.6) | 8(7.8) | |
| 激素受体检测[例(%)] | |||
| ER或PR阳性 | 93(43.9) | 44(42.7) | |
| ER和PR均阴性 | 117(55.2) | 59(57.3) | |
| 不明 | 2(0.9) | 0 | |
| 无病生存期[例(%)]例数 | 195 | 88 | |
| ≤2年 | 117(55.2) | 53(51.5) | |
| >2年 | 78(36.8) | 35(34.0) | |
| 转移部位[例(%)] | |||
| 肝 | 81(38.2) | 42(40.8) | |
| 肺 | 89(42.0) | 44(42.7) | |
| 骨 | 60(28.3) | 36(35.0) | |
| 淋巴结 | 167(78.8) | 80(77.7) | |
| 其他 | 42(19.8) | 21(20.4) | |
| 内脏转移(肺或肝)[例(%)] | |||
| 有 | 142(67.0) | 70(68.0) | |
| 无 | 70(33.0) | 33(32.0) | |
注:KPS为功能状态评分;HER2为人表皮生长因子受体2; IHC为免疫组织化学;FISH为荧光原位杂交技术;ER为雌激素受体;PR为孕激素受体
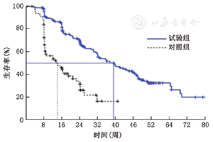
试验组全治疗期(赛普汀联合化疗阶段+赛普汀单药维持治疗阶段)和对照组化疗期进行对比,结果显示两组中位PFS为39.1周(95%CI,32~48)与14.0周(95%CI,8~21),HR=0.24(95%CI,0.16~0.36),P<0.000 1(图1)。
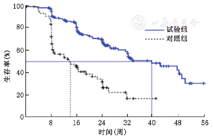
试验组赛普汀联合化疗阶段(不含赛普汀单药维持治疗阶段)和对照组化疗期进行对比,结果显示两组中位PFS为39.6周(95%CI,31~49)与14.0周(95%CI,8~21),HR=0.24(95%CI,0.16~0.37),P<0.000 1(图2)。因此,试验组与对照组相比中位PFS显著延长,疾病进展风险显著降低。
进一步分析受试者PFS的影响因素,对"ER或PR(任一阳性与均阴性)","无病生存期(≤2年与>2年)","内脏转移(有与无)"以及"年龄"进行了多因素分析。结果表明,无论是试验组全治疗期与对照组化疗期对比,还是试验组赛普汀联合化疗阶段与对照组化疗期进行对比,"无病生存期(≤2年与>2年)"对中位数PFS的影响差异均有统计学意义(表2和表3)。
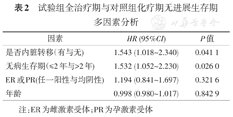
试验组全治疗期与对照组化疗期无进展生存期多因素分析
试验组全治疗期与对照组化疗期无进展生存期多因素分析
| 因素 | HR (95%CI) | P值 |
|---|---|---|
| 是否内脏转移(有与无) | 1.543 (1.018~2.340) | 0.041 1 |
| 无病生存期(≤2年与>2年) | 1.532 (1.052~2.230) | 0.026 0 |
| ER或PR(任一阳性与均阴性) | 1.194 (0.841~1.697) | 0.321 6 |
| 年龄 | 0.998 (0.980~1.017) | 0.842 9 |
注:ER为雌激素受体;PR为孕激素受体
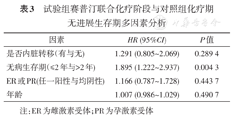
试验组赛普汀联合化疗阶段与对照组化疗期无进展生存期多因素分析
试验组赛普汀联合化疗阶段与对照组化疗期无进展生存期多因素分析
| 因素 | HR (95%CI) | P值 |
|---|---|---|
| 是否内脏转移(有与无) | 1.291 (0.805~2.069) | 0.289 4 |
| 无病生存期(≤2年与>2年) | 1.895 (1.222~2.937) | 0.004 3 |
| ER或PR(任一阳性与均阴性) | 1.166 (0.787~1.728) | 0.443 7 |
| 年龄 | 1.007 (0.986~1.029) | 0.490 7 |
注:ER为雌激素受体;PR为孕激素受体
试验组与对照组相比,ORR和DCR均显著提高(表4),两组ORR为46.7%比18.45% (P<0.000 1),DCR为79.72%比45.63% (P<0.000 1)。
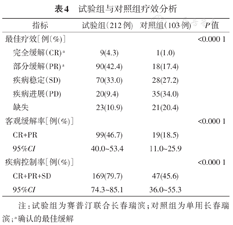
试验组与对照组疗效分析
试验组与对照组疗效分析
| 指标 | 试验组(212例) | 对照组(103例) | P值 | |
|---|---|---|---|---|
| 最佳疗效[例(%)] | <0.000 1 | |||
| 完全缓解(CR)a | 9(4.3) | 1(1.0) | ||
| 部分缓解(PR)a | 90(42.4) | 18(17.4) | ||
| 疾病稳定(SD) | 70(33.0) | 28(27.2) | ||
| 疾病进展(PD) | 20(9.4) | 35(34.0) | ||
| 缺失 | 23(10.9) | 21(20.4) | ||
| 客观缓解率[例(%)] | <0.000 1 | |||
| CR+PR | 99(46.7) | 19(18.5) | ||
| 95%CI | 40.0~53.4 | 11.0~25.9 | ||
| 疾病控制率[例(%)] | <0.000 1 | |||
| CR+PR+SD | 169(79.7) | 47(45.6) | ||
| 95%CI | 74.3~85.1 | 36.0~55.3 | ||
注:试验组为赛普汀联合长春瑞滨;对照组为单用长春瑞滨;a确认的最佳缓解
中性粒细胞减少和白细胞减少在试验组和对照组发生率均较高,总体发生率(任何级别)两组接近(中性粒细胞减少:92.4%比88.8%,白细胞减少:91.6%比87.9%),3~4级发生率试验组高于对照组(中性粒细胞减少:73.8%比56.1%,白细胞减少:61.8%比47.7%)。红细胞减少总体发生率试验组高于对照组(65.3%比49.5%),3~4级发生率接近(9.3%比7.5%)。试验组与对照组相比,发热(51.6%比20.6%)和寒战(19.1%比0.9%)的发生率仍较高,但赛普汀相关的以发热、寒战为临床症状的输注反应多发生在首次给药,且绝大部分为1~2级的轻度反应。消化系统相关AE中恶心呕吐发生率最高,试验组与对照组为29.3%比26.2%,绝大多数为1~2级。肝胆系统相关AE中转氨酶升高发生率最高,试验组与对照组为20.4%比18.7%。呼吸道感染发生率在试验组较对照组高(20.4%比14.0%),但基本均为1~2级。试验组与对照组发生率≥5%不良事件总结见表5。
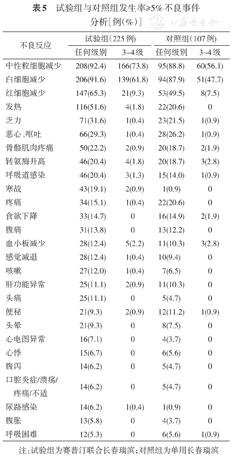
试验组与对照组发生率≥5%不良事件分析[例(%)]
试验组与对照组发生率≥5%不良事件分析[例(%)]
| 不良反应 | 试验组(225例) | 对照组(107例) | ||
|---|---|---|---|---|
| 任何级别 | 3~4级 | 任何级别 | 3~4级 | |
| 中性粒细胞减少 | 208(92.4) | 166(73.8) | 95(88.8) | 60(56.1) |
| 白细胞减少 | 206(91.6) | 139(61.8) | 94(87.9) | 51(47.7) |
| 红细胞减少 | 147(65.3) | 21(9.3) | 53(49.5) | 8(7.5) |
| 发热 | 116(51.6) | 4(1.8) | 22(20.6) | 0 |
| 乏力 | 71(31.6) | 1(0.4) | 23(21.5) | 1(0.9) |
| 恶心、呕吐 | 66(29.3) | 1(0.4) | 28(26.2) | 1(0.9) |
| 骨骼肌肉疼痛 | 50(22.2) | 2(0.9) | 20(18.7) | 2(1.9) |
| 转氨酶升高 | 46(20.4) | 4(1.8) | 20(18.7) | 3(2.8) |
| 呼吸道感染 | 46(20.4) | 3(1.3) | 15(14.0) | 1(0.9) |
| 寒战 | 43(19.1) | 2(0.9) | 1(0.9) | 0 |
| 疼痛 | 34(15.1) | 1(0.4) | 22(20.6) | 0 |
| 食欲下降 | 33(14.7) | 0 | 16(14.9) | 2(1.9) |
| 腹痛 | 31(13.8) | 0 | 13(12.2) | 0 |
| 血小板减少 | 28(12.4) | 5(2.2) | 11(10.3) | 3(2.8) |
| 感觉减退 | 28(12.4) | 1(0.4) | 10(9.4) | 0 |
| 咳嗽 | 27(12.0) | 1(0.4) | 7(6.5) | 0 |
| 肝功能异常 | 25(11.1) | 2(0.9) | 11(10.3) | 0 |
| 头痛 | 25(11.1) | 0 | 5(4.7) | 0 |
| 便秘 | 21(9.3) | 2(0.9) | 12(11.2) | 1(0.9) |
| 头晕 | 21(9.3) | 0 | 8(7.5) | 0 |
| 心电图异常 | 16(7.1) | 0 | 4(3.7) | 0 |
| 心悸 | 15(6.7) | 0 | 6(5.6) | 0 |
| 腹泻 | 14(6.2) | 0 | 5(4.7) | 0 |
| 口腔炎症/溃疡/疼痛/不适 | 14(6.2) | 0 | 5(4.7) | 0 |
| 尿路感染 | 14(6.2) | 1(0.4) | 1(0.9) | 0 |
| 腹胀 | 13(5.8) | 0 | 4(3.7) | 0 |
| 呼吸困难 | 12(5.3) | 0 | 6(5.6) | 1(0.9) |
注:试验组为赛普汀联合长春瑞滨;对照组为单用长春瑞滨
在本研究中,共有12例患者发生SAE,其中试验组6例(发生率2.7%),对照组6例(发生率5.6%),组间差异无统计学意义。经研究者判断,试验组SAE中,中性粒细胞减少伴发热、粒细胞缺乏症伴肠道感染与长春瑞滨和(或)赛普汀用药肯定有关,SAE转归均为症状消失且无后遗症;双侧肺部感染与长春瑞滨和(或)赛普汀用药可能有关,SAE转归为症状消失且无后遗症。对照组SAE中,肠梗阻、血小板下降3级合并粒细胞缺乏症伴感染、食欲下降3级与长春瑞滨用药肯定有关,SAE转归为症状消失且无后遗症。
治疗过程中,出现心悸症状的患者两组共21例,其中试验组15例,发生率6.7%,对照组6例,发生率5.6%,两组间差异无统计学意义;发生心电图异常两组共20例,其中试验组16例,发生率7.1%,对照组4例,发生率3.7%,均为1~2级,未影响用药。治疗过程中监测LVEF降低至<50%的情况在试验组发生了3例,对照组发生了2例。试验组3例中2例LVEF较基线下降幅度较大(分别为36.9%和34.7%),但经暂停药物后均回升至>50.0%,另1例较基线下降幅度小(7.5%),监测其自行恢复至>50.0%。对照组2例LVEF下降均发生在序贯单药赛普汀治疗期,较基线下降幅度较小(分别为11.3%和13.2%),因肿瘤进展结束用药后随访恢复至>50%。
在HER2阳性复发转移性乳腺癌治疗中,赫赛汀联合紫杉醇/多西紫杉醇的两项Ⅲ期临床研究结果[1,2],确立了赫赛汀联合紫杉类一线治疗的地位,然而,在临床治疗实践中,越来越多晚期乳腺癌患者在早期治疗阶段应用过紫杉类。本研究评价了中国自主研发的创新药物重组抗HER2人源化单克隆抗体(赛普汀)联合长春瑞滨用于既往紫杉类治疗后,但未经抗HER2靶向治疗的HER2阳性晚期乳腺癌患者的疗效和安全性。
本试验主要终点PFS分析结果表明,无论试验组整个治疗期(赛普汀联合化疗+赛普汀单药维持)与对照组化疗,还是试验组赛普汀联合化疗与对照组化疗,中位PFS均显著延长,疾病进展风险显著降低(39.1周与14.0周、39.6周与14.0周,均P<0.000 1)。次要终点肿瘤ORR以及DCR结果提示,试验组赛普汀联合化疗与对照组化疗均显著提高(ORR为46.7%与18.45%,P<0.000 1;DCR为79.72%与45.63%,P<0.000 1)。因此,在长春瑞滨化疗基础上联合应用抗HER2抗体药物赛普汀相比化疗单药,能够显著延长PFS,显著提高ORR和DCR。此外,对照组化疗出现肿瘤进展或毒性不耐受时序贯接受赛普汀单药治疗的患者,部分也表现出疗效,疾病控制时间得到延长。
既往文献报道长春瑞滨联合曲妥珠单抗治疗的总体有效率44%~86%,一线治疗为51%~86%,中位PFS约9~10个月[3,4,5,6,7,8,9],这些研究多为小样本量的Ⅱ期研究,2009年本研究启动时,同类Ⅲ期研究尚未见结果报道。一项纳入178例受试者的回顾性研究中,赫赛汀联合长春瑞滨一线治疗TTP为10个月,ORR为57% [10]。HERNATA研究是一项前瞻性Ⅲ期研究,结果显示141例患者接受赫赛汀联合长春瑞滨一线治疗中位TTP为15.3个月,ORR为59.3%[11]。本研究结果与上述研究结果进行对比可见,本研究纳入受试者非全部为一线治疗,包括了一线至四线治疗的患者,接受赛普汀联合长春瑞滨治疗PFS近10个月,与前述一线治疗回顾性研究中赫赛汀联合长春瑞滨PFS相当,赛普汀联合长春瑞滨治疗ORR为46.7%,略低于赫赛汀联合长春瑞滨用于一线治疗的ORR。
本研究发生率较高的AE是中性粒细胞减少、白细胞减少和红细胞减少,上述血液学毒性虽试验组较对照组有所增加,但差异无统计学意义,且均是短暂、可逆和非累积性的,通过长春瑞滨剂量调整以及使用对症治疗药物可恢复,此结果与国内外同类研究一致[10,12]。最常见的与赛普汀用药相关的AE是输注反应,表现为发热和寒战,绝大多数发生在首次用药时,为轻度反应,易耐受,不治疗或对症治疗后症状可完全消失,不影响后续药物使用。本研究无超敏反应、血管性水肿或肺毒性等严重和致命的输注反应。国内外诸多相关研究结果均显示抗HER2单抗药物存在心脏毒性[11,13],因此赛普汀相关心脏毒性也是本研究安全性评价的重点,本试验期间未出现心功能不全或心力衰竭等严重心脏毒性。共5例患者治疗中LVEF降低至<50%,均自行或经暂停药物后回升至>50%。总体上说,在长春瑞滨基础上联合赛普汀治疗无显著性增加的严重毒性。
来自中国的真实世界研究[14]表明,在中国不同地区赫赛汀应用存在很大差异,在资源匮乏的地区,即使到了2015年,患者接受治疗的比例仍低,直至2017年赫赛汀进入国家医保用药,HER2阳性乳腺癌患者能够使用抗HER2治疗的比例才有所提高。本试验在研究期间(2009年至2013年)为中国HER2阳性乳腺癌患者提供了更多靶向治疗机会,研究结果显示注射用重组抗HER2人源化单克隆抗体(赛普汀)联合长春瑞滨具有显著疗效和良好安全性,是紫杉类治疗后HER2阳性晚期乳腺癌的优选方案。
参与研究的26家中心所有研究者:解放军总医院第五医学中心(原军事医学科学院附属医院)(江泽飞);中国医学科学院肿瘤医院(徐兵河);北京大学肿瘤医院(任军,邸立军);浙江省肿瘤医院(王晓稼);解放军总医院第一医学中心(焦顺昌,杨俊兰);天津市肿瘤医院(佟仲生);福建省肿瘤医院(陈强,郑弘宇,刘健);江苏省肿瘤医院(冯继锋,孙蔚莉);中山大学肿瘤防治中心(刘冬耕);广西医科大学附属肿瘤医院(于起涛);中国医科大学附属第一医院(刘云鹏);四川大学华西医院(郑鸿,鄢希);南方医科大学南方医院(罗荣城);湖南省肿瘤医院(汪安兰,欧阳取长);空军军医大学西京医院(原第四军医大学第一附属医院)(刘文超);河北医科大学第四医院(刘巍,左静);天津市人民医院(李维廉,姚嫱);华中科技大学同济医学院附属同济医院(于世英);解放军第九〇〇医院(原福州总医院)(欧阳学农,陈曦);解放军第九六〇医院(原济南军区总医院)(王宝成,毕经旺);上海市第一人民医院(王理伟,李琦);蚌埠医学院附属医院(郑荣生);中南大学湘雅二医院(胡春宏);重庆市肿瘤医院(吕钢,曾晓华);安徽医科大学第一附属医院(孙国平);四川省人民医院(刘锦平)
感谢研究专家组孙燕院士、宋三泰教授、郭亚军教授、石远凯教授、姚晨教授为本研究所做的工作
所有作者均声明不存在利益冲突

























