
研究黄连素对脓毒症大鼠心肌损伤和心功能障碍的影响及其作用机制。
将实验SD大鼠采用随机数字表法分为3组:脓毒症组(LPS组)、黄连素干预组(Ber组)、对照组。LPS组给予腹腔注射LPS(10 mg/kg);Ber组实验前3 d开始灌胃Ber(50 mg/kg,每日1次),第3天灌胃1 h后腹腔注射LPS;对照组(Con组),分别给予双蒸水(2 ml/100 g)灌胃和生理盐水(1 ml/100 g)腹腔注射。LPS组和Ber组每组又分为3个亚组(n=6),分别于LPS注射后6、24和48 h进行后续实验(其中48 h亚组大鼠在24 h时给予灌胃Ber/生理盐水1次)。监测大鼠左心室收缩压(LVSP)、左心室舒张末期压(LVEDP)、左心室压力最大变化速率(±dp/dtmax);ELISA法检测大鼠血浆中心肌损伤标志物心肌肌钙蛋白T(cTnT)水平以及心肌组织肿瘤坏死因子(TNF)-α和白细胞介素(IL)-1β水平;取心肌组织行HE染色,观察心肌损伤情况;Western印迹法检测大鼠心肌组织Toll样受体4(TLR4)蛋白表达情况;检测大鼠心肌细胞核内p65蛋白水平反映核因子(NF)-κB活化程度。用Pearson相关系数法分析各因素间的相关性。
LPS 6、24和48 h组大鼠的LVSP、+dp/dtmax和-dp/dtmax均明显低于对照组(均P<0.05),LVEDP均高于对照组(均P<0.05),Ber 48 h组大鼠LVSP高于LPS 48 h组(均P<0.05),Ber 6、24和48 h组大鼠的±dp/dtmax均高于相应LPS组(均P<0.05),Ber 6、24和48 h组大鼠的LVEDP均低于相应LPS组(均P<0.05)。LPS 6、24和48 h组大鼠cTnT浓度明显高于对照组(均P<0.05),Ber 6、24和48 h组大鼠cTnT浓度均低于相应LPS组(均P<0.05)。与对照组相比,LPS 6 h组大鼠心肌细胞肿胀肥大,心肌细胞纤维部分断裂,可见淋巴细胞和中性粒细胞浸润,LPS 24 h组最为严重。Ber 6、24和48 h组大鼠心肌细胞的损伤表现及炎症细胞浸润情况较相应LPS组轻。LPS 6 h、LPS 24 h和LPS 48 h组大鼠心肌组织上TLR4蛋白表达、心肌细胞核内p65蛋白浓度、心肌组织TNF-α、IL-1β含量均较对照组升高(均P<0.05),Ber 6 h、Ber 24 h、Ber 48 h组大鼠上述指标较相应LPS组低(均P<0.05)。
Ber可改善脓毒症大鼠心肌损伤和心功能,机制考虑与其抑制LPS诱导的TLR4/NF-κB信号通路激活,降低TNF-α、IL-1β等炎症因子表达有关。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
脓毒症患者病死率虽然已显著下降,但仍高达20%[1]。我国每年有413万例成人脓毒症患者,每年相关医疗费用支出高达141亿美元[2]。40%~60%的脓毒性休克患者合并脓毒性心功能障碍,又称为脓毒性心肌病(sepsis cardiomyopathy,SCM)[3],其病死率则显著升高。有效防治SCM是降低脓毒症患者病死率的关键。SCM的发病机制目前尚未完全阐明,寻找防治SCM的有效方法是当前重症医学的任务之一。黄连素(Ber),又名小檗碱,是从传统中药黄连和黄柏中分离得到的异喹啉类生物碱。近年研究表明,黄连素能提高脓毒症小鼠的存活率[4]。黄连素对SCM是否有保护作用及其机制如何,目前少见报道。本研究探讨黄连素对SCM的作用及其机制。
健康雄性SD大鼠42只,体重250~300 g,由广东省医学动物实验中心提供[动物合格证号:SCXK(粤)2013-0002]。饲养于12 h光照/12 h无光照环境,实验室环境温度控制在(25±1)℃,相对湿度50%~70%,5只/笼饲养,自由进食及饮水,适应饲养1周。实验前禁食12 h。
大肠杆菌脂多糖(LPS)和盐酸小檗碱(Ber)(美国Sigma-Aldrich公司),大鼠血浆心肌肌钙蛋白T(cTnT)ELISA试剂盒、大鼠心肌组织肿瘤坏死因子(TNF)-α ELISA试剂盒、大鼠心肌组织IL-1β ELISA试剂盒(武汉华美生物公司),细胞核提取试剂盒、TransAMTM核因子(NF)-κB p65转录因子检测试剂盒(美国Active Motif公司),兔抗鼠Toll样受体4(TLR4)多克隆抗体(美国Santa Cruz公司)。BL-420F生物信号采集与分析系统(成都泰盟科技公司)。
采用数字随机表法随机将大鼠分为3组,即对照组(Con组)(n=6)、脓毒症模型组(LPS组)(n=18)和黄连素干预组(Ber组)(n=18)。Ber组大鼠实验前3 d开始给予小檗碱(50 mg/kg)灌胃,每日1次,对照组大鼠予同等剂量双蒸水(2 ml/100 g)灌胃。在第3天灌胃后1 h,给Ber组和LPS组大鼠腹腔注射LPS 10 mg/kg建立脓毒症大鼠模型,对照组大鼠腹腔注射生理盐水1 ml/100 g,观察时间点为腹腔注射LPS或生理盐水后6、24和48 h。根据观察的时间截点,LPS组又分为3个亚组,即LPS 6 h组、LPS 24 h组和LPS 48 h组。Ber组亦分为3个亚组,即Ber 6 h组、Ber 24 h组、Ber 48 h组,其中Ber 48 h组大鼠在腹腔注射LPS 24 h时给予小檗碱(50 mg/kg)灌胃1次。由于对照组未加任何干预措施,随着时间进展,各检测指标无显著差异,因此,以对照组腹腔注射生理盐水6 h作为不同时间点的对照。每个亚组大鼠数量均为6只。
在腹腔注射LPS或生理盐水后6、24和48 h,腹腔注射3%戊巴比妥钠(0.2 ml/100 g),达满意麻醉效果后固定于操作台上。行气管插管接呼吸机辅助通气。先将PT-100压力感受器一端连到BL-420F生物机能实验系统的1通道,另一端与PE50聚乙烯管相连。接好呼吸机后,分离右颈总动脉,给予颈总动脉插管并将插管前端送入左心室腔内,将导管结扎固定好。待波形稳定后,分别记录左心室收缩峰压(LVSP)、左心室舒张末期压(LVEDP)、左心室收缩时室内压最大上升速率(+dp/dtmax)和左心室舒张时室内压最大下降速率(-dp/dtmax)。
实验四各组大鼠右颈总动脉心室插管血流动力学监测完毕后,将PE50管末端连接注射器,每只大鼠抽取2 ml血液,4 ℃条件下3 000 r/min离心10 min,吸取上清液,分装于EP管中并保存于-20 ℃冰箱中备用。采用ELISA法检测各组大鼠cTnT浓度。具体实验操作步骤按照试剂盒说明书进行。
各组采血结束后用颈椎脱臼法处死大鼠,迅速摘取心肌组织,生理盐水冲洗干净后吸水纸吸净表面水分。然后取心尖部分的一小块心肌组织,经4%中性甲醛溶液固定,常规取材、脱水、石蜡包埋、连续切片,切片厚度4 μm,HE染色,光学显微镜观察大鼠心肌细胞损伤和炎症浸润的情况。
将实验六中摘取的剩余心肌组织分成2部分,一部分制成组织裂解液:取100 mg心肌组织剪碎后制成组织匀浆,-20 ℃过夜,反复冻融2次,破坏心肌细胞膜,然后将组织匀浆置于4 ℃,5 000 g离心5 min,吸取上清,-80 ℃保存备用。严格按照胞质蛋白试剂盒说明书提取蛋白。并按照常规Western印迹法检测大鼠心肌组织上TLR4蛋白的表达。另一部分心肌组织液氮速冻,-80 ℃冰箱保存备用。
本实验采用核蛋白提取试剂盒抽提实验六中分离心肌组织的核蛋白,具体步骤参照试剂盒说明书。参照TransAM NF-κB p65试剂盒说明书操作,取胞核提取物2.5 μg与96孔板孵育(96孔板中包被有NF-κB结合位点序列5′-GGGACTTTCC-3′),只有胞核提取物内活化的p65才能与之结合[5]。具体操作步骤参照文献[6]。
取实验六中摘取的心肌组织,制成匀浆,采用ELISA法检测各组大鼠心肌组织炎症因子TNF-α和IL-1β蛋白含量,具体步骤按照试剂盒说明书进行。
采用SPSS 20.0对实验数据进行统计分析。符合正态分布的计量资料以 ±s表示,3组间比较采用单因素方差分析,两两比较采用最小显著性差异法。用Pearson相关系数法分析各因素间的相关性。检验水准值取双侧α=0.05。
±s表示,3组间比较采用单因素方差分析,两两比较采用最小显著性差异法。用Pearson相关系数法分析各因素间的相关性。检验水准值取双侧α=0.05。
LPS 6、24和48 h组大鼠的LVSP、+dp/dtmax和-dp/dtmax均明显低于对照组(均P<0.05),LVEDP均高于对照组(均P<0.05),提示脓毒症大鼠SCM模型建立成功。Ber 48 h组大鼠LVSP高于LPS 48 h组(P<0.05),Ber 6、24和48 h组大鼠的+dp/dtmax、-dp/dtmax则分别高于LPS 6、24和48 h组(均P<0.05),Ber 6、24和48 h组大鼠的LVEDP分别低于LPS 6、24和48 h组(均P<0.05)。提示Ber可抑制LPS诱导的SCM大鼠心肌收缩和舒张功能下降,改善心功能(表1)。
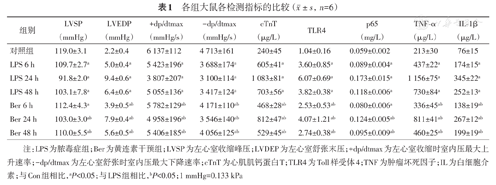
各组大鼠各检测指标的比较( ±s,n=6)
±s,n=6)
各组大鼠各检测指标的比较(±s,n=6)
| 组别 | LVSP(mmHg) | LVEDP(mmHg) | +dp/dtmax(mmHg/s) | -dp/dtmax(mmHg/s) | cTnT(μg/L) | TLR4 | p65(mg/L) | TNF-α(μg/L) | IL-1β(μg/L) |
|---|---|---|---|---|---|---|---|---|---|
| 对照组 | 119.0±3.1 | 2.2±0.4 | 6 137±112 | 4 713±161 | 240±45 | 1.04±0.16 | 0.059±0.002 | 213±30 | 76±15 |
| LPS 6 h | 109.7±2.7a | 5.0±0.4a | 5 423±196a | 3 688±174a | 605±41a | 3.60±0.85a | 0.089±0.004a | 437±22a | 174±15a |
| LPS 24 h | 91.8±2.0a | 9.4±0.6a | 3 807±207a | 3 100±114a | 1 083±81a | 6.07±0.69a | 0.173±0.015a | 1 156±75a | 345±22a |
| LPS 48 h | 103.1±7.8a | 6.4±0.6a | 5 055±136a | 3 417±124a | 703±56a | 3.82±0.38a | 0.118±0.006a | 730±84a | 252±13a |
| Ber 6 h | 112.4±4.3a | 3.9±0.5ab | 5 782±129ab | 4 171±110ab | 468±28ab | 2.53±0.53ab | 0.080±0.006a | 336±45ab | 138±19ab |
| Ber 24 h | 103.0±3.0ab | 7.9±0.4ab | 4 958±196ab | 3 546±140ab | 812±47ab | 4.07±1.21ab | 0.124±0.005ab | 811±41ab | 267±12ab |
| Ber 48 h | 110.0±5.5ab | 5.6±0.5ab | 5 406±185ab | 4 056±125ab | 529±45ab | 2.74±0.38ab | 0.095±0.009ab | 460±25ab | 199±19ab |
注:LPS为脓毒症组;Ber为黄连素干预组;LVSP为左心室收缩峰压;LVDEP为左心室舒张末压;+dp/dtmax为左心室收缩时室内压最大上升速率;-dp/dtmax为左心室舒张时室内压最大下降速率;cTnT为心肌肌钙蛋白T;TLR4为Toll样受体4;TNF为肿瘤坏死因子;IL为白细胞介素;与Con组相比,aP<0.05;与LPS组相比,bP<0.05;1 mmHg=0.133 kPa
LPS 6、24和48 h组大鼠cTnT浓度明显高于对照组(均P<0.05),以LPS 24 h组为最高,结合大鼠血流动力学监测结果,提示大鼠SCM建立成功。Ber 6、24和48 h组大鼠cTnT浓度分别低于相应LPS组(均P<0.05)。提示Ber可减轻LPS诱导的SCM大鼠心肌损伤(表1)。
HE染色结果显示与对照组相比,LPS 6 h组大鼠心肌细胞肿胀肥大,心肌细胞纤维部分断裂,可见淋巴细胞和中性粒细胞浸润,LPS 24 h组大鼠上述改变更加明显,LPS 24 h组大鼠上述心肌细胞损伤表现和炎症细胞的浸润则最为严重。Ber 6、24和48 h组大鼠心肌细胞的损伤表现及炎症细胞浸润情况则较相应时间点的LPS各亚组轻。提示Ber可改善LPS诱导的SCM大鼠心肌细胞损伤和炎症细胞浸润(图1)。
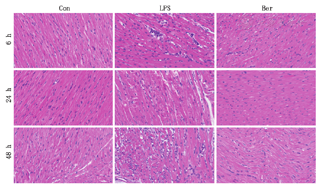
注:Con为对照组;LPS为脓毒症组;Ber为黄连素干预组
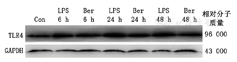
LPS 6 h、LPS 24 h和LPS 48 h组大鼠心肌组织上TLR4蛋白表达较对照组升高(均P<0.05),Ber 6 h、Ber 24 h、Ber 48 h组大鼠模型心肌组织TLR4蛋白表达水平较相应LPS组降低(均P<0.05)。提示Ber可下调LPS诱导的大鼠模型心肌组织TLR4表达水平(图2、表1)。
LPS 6、24和48 h组大鼠心肌细胞核内p65蛋白浓度较对照组高(均P<0.05),Ber 24 h和48 h组大鼠心肌细胞核内p65蛋白浓度较相应LPS组低(均P<0.05)。提示Ber可部分抑制LPS诱导的SCM大鼠心肌细胞NF-κB信号活化(表1)。
LPS组大鼠模型心肌组织TNF-α、IL-1β水平在模型建立6、24和48 h时与对照组相比均升高(均P<0.05),Ber组大鼠心肌组织TNF-α、IL-1β水平在上述时间点均较LPS组降低(均P<0.05)(表1)。
Pearson相关分析结果显示,对照组cTnT与心肌组织TLR4/NF-κB信号通路关键信号分子(TLR4、p65、TNF-α、IL-1β)均为正常水平,相互之间不呈现显著相关。LPS刺激后,大鼠cTnT与心肌组织TLR4/NF-κB信号通路关键信号分子(TLR4、p65、TNF-α、IL-1β)相对表达水平上升且呈正相关(均P<0.001,相关系数分别为:0.91,0.97,0.97, 0.96)。Ber组大鼠cTnT与心肌组织TLR4/NF-κB信号通路的关键信号分子(TLR4、p65、TNF-α、IL-1β)相对表达水平低于LPS组,相互之间呈正相关(r=0.88、0.94、0.97、0.98,均P<0.001)。
SCM是脓毒症相关器官功能障碍重要组成部分。1984年Parker等[7]首次报道了脓毒症引起的心肌抑制,此后虽有大量研究尝试揭示SCM的发病机制[8,9,10],但SCM的治疗仍以支持治疗为主,尚缺乏针对性治疗药物及手段。近年研究发现祖国传统中药Ber能提高脓毒症小鼠的存活率,另有研究发现黄连素能激活α2肾上腺素受体并诱导细胞因子的表达[11,12],王一阳等[13]发现Ber可抑制LPS诱导的心功能障碍,但与其激活α2肾上腺素受体无关。因此有必要针对Ber改善SCM的机制进行深入研究。
Con:对照组;LPS:脓毒症模型组;Ber:黄连素干预组;TLR4为Toll样受体4;GAPDH为甘油醛-3-磷酸脱氢酶(内参)
SCM制模尚无明确统一的评定标准,通过检测脓毒症模型心功能和心肌损伤标志物,观察心肌组织形态学改变,综合评价SCM模型是否成功[14]。实验中我们采用给大鼠腹腔注射LPS 10 mg/kg的方法建立脓毒症模型,发现大鼠在建模后6 h时即开始出现反映心肌收缩功能的指标LVSP、+dp/dtmax以及反映心肌舒张功能的指标-dp/dtmax下降,LVEDP升高,血浆心肌损伤标志物cTnT升高,心肌组织HE染色可见心肌细胞损伤和炎性细胞浸润,上述改变在模型建立后24 h最为严重,根据文献报道[14]可评定SCM模型大鼠构建成功。Ber干预的脓毒症大鼠心脏收缩和舒张功能改善,cTnT水平降低,心肌细胞损伤和炎症细胞浸润减轻。提示Ber对LPS诱导的SCM模型大鼠心肌损伤和心功能具有保护作用,机制考虑与其抑制炎症反应有关。
脓毒症时TLR4特异识别LPS,TLR4的活化最终表现在mRNA和蛋白水平上激活下游NF-κB信号通路[15],诱导大量活性氮类物质、促炎细胞因子IL-1、IL-6和TNF-α等的过度释放[16],这些级联反应与脓毒症所诱导的心肌损伤密切相关。有研究表明Ber可通过抑制肺组织细胞IκB的降解,减少细胞因子TNF-α、IL-1β的释放[17]。为进一步探讨Ber对脓毒症大鼠SCM产生保护作用的机制,本研究检测了各组大鼠心肌细胞上TLR4受体、NF-κB信号活化情况及下游炎症因子TNF-α、IL-1β的水平。发现Ber干预可使脓毒症大鼠心肌组织TLR4受体表达水平、心肌细胞NF-κB信号活化程度以及心肌组织TNF-α、IL-1β表达水平降低。对3组大鼠cTnT水平和心肌组织TLR4、TNF-α、IL-1β水平以及心肌细胞核内p65水平的相关性分析显示cTnT与它们呈正相关。据此笔者推测Ber可能通过抑制TLR4/NF-κB信号通路,减轻炎症因子对大鼠模型心肌损伤从而对SCM起保护作用,其是否同时还通过其他机制对SCM起保护作用仍需进一步研究。
综上,本研究显示Ber可减轻脓毒症大鼠模型的心肌损伤,增强心肌收缩和舒张功能从而改善SCM,其机制考虑与Ber抑制脓毒症诱发的TLR4/NF-κB信号通路激活,降低TNF-α、IL-1β等炎症因子的表达水平,减轻对心肌细胞的损伤有关。Ber有望用于治疗SCM。本研究存在一些遗憾,如没有检测Ber血药浓度,Ber是否还通过调控其他机制比如对心肌线粒体动力学影响等起到改善SCM的作用尚不明确,均有待进一步研究证实。
广东省医学科研基金(B2018029)和广州市医药卫生科技项目(20181A011019)于广州市红十字会医院获得,特此志谢!
所有作者均声明不存在利益冲突

























