
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
生物类似药是指在质量、安全性和有效性方面与已获准上市的参照药具有相似性的治疗性生物制品[1]。生物类似药作为生物制剂,具有分子量大、空间结构和理化性质复杂、异质性大等特点,相较于化学仿制药,生物类似药对生产工艺要求更高、对生产过程更加敏感、研发与生产投入的周期也更长。因此生物类似药要做到与参照药完全一致几乎是不可能的,这也正是生物药的仿制品只能被称为生物类似药(biosimilar),而不能被称为生物仿制药(biogeneric)的原因。由于生物类似药在药理特性、生物活性、安全性、有效性等方面与参照药相似,而价格相对原研药低,更具有可及性优势,对减轻个人与国家的医疗保健经济负担,让更多患者获益,具有积极意义。
随着越来越多的原研生物药专利到期,全球生物类似药逐渐呈现蓬勃发展之势。欧洲药品管理局(EMA)于2006年批准上市了全球第一款生物类似药。截止2019年5月29日,全球生物类似药研发处于活跃状态的有718个、上市121个、批准15个、注册阶段31个、Ⅲ期临床试验阶段72个、Ⅱ期临床试验阶段8个、Ⅰ期临床试验阶段75个,还有386个处于研发阶段[2]。我国在生物类似药领域虽然起步较晚,但发展迅速。2015年,国家药品监督管理局(NMPA)颁布了《生物类似药研发与评价技术指导原则(试行)》(下称《指导原则》)[1],截止2019年5月29日,我国更是以251个在研生物类似药的数量领跑全球[2]。
我国对于生物类似药的上市过程采取逐步递进的路径,除了药代动力学(PK)/药效动力学(PD)对比研究证明两者的相似性之外,还需进行临床有效性的对比研究[1]。截止2020年3月,我国已有4个生物类似药获批上市(表1)。

我国已上市的4种生物类似物
我国已上市的4种生物类似物
| 生物类似药(商品名) | 参照药(商品名) | 获批时间 |
|---|---|---|
| 利妥昔单抗注射液(汉利康®)[3] | 利妥昔单抗(美罗华®) | 2019年2月22日 |
| 阿达木单抗注射液(格乐立®)[4] | 阿达木单抗(修美乐®) | 2019年11月7日 |
| 阿达木单抗注射液(安健宁®)[5] | 阿达木单抗(修美乐®) | 2019年12月6日 |
| 贝伐珠单抗注射液(安可达®)[6] | 贝伐珠单抗(安维汀®) | 2019年12月9日 |
注:利妥昔单抗注射液(汉利康®)上海复宏汉霖生物制药有限公司(中国)生产,国药准字S20190021;利妥昔单抗(美罗华®)罗氏制药有限公司(瑞士)生产,国药准字J20170034、J20170005;阿达木单抗注射液(格乐立®)广东百奥泰生物制药股份有限公司(中国)生产,国药准字S20190038;阿达木单抗注射液(安健宁®)浙江海正生物制药有限公司(中国)生产,国药准字S20190043;阿达木单抗(修美乐®)艾伯维有限公司(美国)生产,国药准字S20191006;贝伐珠单抗注射液(安可达®)齐鲁制药有限公司(中国)生产,国药准字S20190040;贝伐珠单抗(安维汀®)罗氏制药有限公司(瑞士)生产,进口药品注册证号S20170035、S20170036
当前,生物类似物在国内刚刚起步,尚缺乏临床应用管理经验,有待在进一步实施过程中不断积累经验。除此之外,在类似药与原研药的互换、适应证外推、安全性监测、处方审核等方面,尚缺乏明确的标准,相关的政策规定还待建立或待逐步完善,给临床应用管理工作带来一定困惑。由中华医学会临床药学分会专家委员会牵头,组织该领域的相关专家,参考国内外政策法规、指南意见及临床研究等,结合我国实际情况,讨论并制定了《生物类似药临床应用管理专家共识(第一版)》,旨在为生物类似药的临床应用管理提供参考。
(1)专家组构成:由以药学专家为主的共识制定组牵头主导,以临床专家为主的咨询专家组提供咨询意见,共同参与制定本《共识》。(2)共识注册:本共识已在国际实践指南注册平台(International Practice Guideline Registry Platform,IPGRP,http://www.guidelines-registry.org)进行注册(注册号为IPGRP-2020CN014)。(3)问题来源:由共识制定组从药学角度拟定问题,经共识制定组与咨询专家组讨论并确定与生物类似药临床应用管理关系密切的4个问题纳入本《共识》。(4)推荐级别:对有循证证据的问题,采用牛津循证医学中心的证据分级水平与推荐级别[7]。对缺少循证证据的问题,采用专家讨论表决的方式。A级(100%一致):所有专家完全达成共识,推荐;B级(75%~99%一致):绝大多数专家达成共识,可推荐;C级(50%~74%一致):多数专家达成共识少数专家存在分歧;D级(<50%一致):未达成共识。
生物类似药研发的总策略是以比对试验为基础,证明生物类似药与参照药(专利到期)在药物特性、安全性与有效性上具有相似性[1]。生物类似药与原研药的相似性可分为两个层次:生物相似性与可互换性,前者是基本要求,后者是更高要求。生物相似性主要基于理化分析、免疫学分析、生物学活性分析(包括体外活性,以及体内的PK/PD特性);可互换性则需要接受更加严格的临床试验,针对特定病种证实生物类似药与参照药在任意给定的患者群体中,都预期具有相同的临床结局[8,9,10]。由于"可互换性"的标准制定,涉及到临床医师、药师、患者、支付方、政策制定者等多方利益的考量与平衡,因此,目前各国或地区对"互换性"的标准不完全相同。我国目前还未建立生物类似药"可互换"的标准,有学者认为,鉴于大分子结构特点难以做到完全一致,进而实现完全互换,现阶段在注册审评环节可暂不设立"可互换的生物类似药"[11]。
生物类似药的临床应用在我国才刚刚起步,已有的研究数据比较有限,考虑到国家对生物类似药与参照药的互换管理还没有具体要求,因此,积极开展RWS并获取真实世界数据,对于推动生物类似药在中国的合理使用有着十分重要的意义。虽然生物类似药在申请上市时已递交了药物安全性的技术资料,但临床前安全性评价不可能全面预测其安全问题,加之Ⅲ期临床试验的病例数有限,研究时间较短,并有严格的纳排标准,研究结果的外推性不满意[12],可能存在某些未被发现的、罕见的、严重的安全问题,有必要待上市后在更大人群样本中进行验证[13]。而RWS是对临床实际产生的真实数据进行系统性收集与分析的研究,具有宽泛的纳入标准和较少的排除标准、较大的样本量数据、较长的观测评估,评价结果的外部真实性好,最接近于真实情况的临床实践[14]。2020年NMPA颁布了《真实世界证据支持药物研发与审评的指导原则(试行)》,对上市后药物利用真实世界数据对药物在真实医疗实践中的效果、安全性、使用情况以及经济学效益等方面进行更全面的评估做出了指导[15]。对生物类似药的临床综合评价需要考虑多种因素,完整的证据链对于建立生物类似药的临床综合评价的标准规范有着重要意义。在国外,部分生物类似药通过了多种临床研究的验证(表2),国外的研究内容为我国生物类似药的临床研究提供了参考,除了头对头和RWS外,还可以通过完善其他临床研究以促进生物类似药的安全使用,鼓励生物类似药在上市后进行RWS和临床综合评价。

部分生物类似药与原研药的比对研究
部分生物类似药与原研药的比对研究
| 靶点 | 参考药(商品名) | 生物类似药(商品名) | 治疗疾病 | 研究类型 | 证据等级 |
|---|---|---|---|---|---|
| G-CSFa | 非格司亭(Neupogen®) | EP2006(Zarzio®)[16] | 化疗后中性粒细胞减少 | Ⅲ期RCTb | 1b |
| TNF-αc | 阿达木单抗(Humira®) | MSB11022[17] | 类风湿关节炎 | Ⅲ期RCTb | 1b |
| BI 695501(Cyltezo®)[18] | 类风湿关节炎 | Ⅲ期RCTb | 1b | ||
| ABP501(Amjevita®)[19] | 银屑病 | Ⅲ期RCTb | 1b | ||
| 英夫利昔单抗(Remicade®) | CT-P13(Inflectra®/Remsima®) | Crohn病、溃疡性结肠炎结、类风湿关节炎、强直性脊柱炎、银屑病[20] | Ⅳ期RCTb | 1b | |
| Crohn病[21] | Ⅲ期RCTb | 1b | |||
| 炎症性肠病[22] | Ⅳ期RCTb | 1b | |||
| 依那西普(Enbrel®) | GP2015[23] | 银屑病 | Ⅲ期RCTb | 1b | |
| HER2d | 曲妥珠单抗(Herceptin®) | ABP 980(KanjintiTM)[24] | 乳腺癌 | Ⅲ期RCTb | 1b |
| SB3[25] | 乳腺癌 | Ⅲ期RCTb | 1b | ||
| CT-P6[26] | 乳腺癌 | Ⅲ期RCTb | 1b | ||
| CD20e | 利妥昔单抗(MabThera®) | CT-P10(Truxima®)[27] | 淋巴瘤 | Ⅲ期RCTb | 1b |
| VEGFf | 贝伐珠单抗(Avastin®) | BEVZ92[28] | 转移性结直肠癌 | 多中心RCTb | 1b |
注:G-CSF为粒细胞集落刺激因子;RCT为随机对照试验;TNF-α为肿瘤坏死因子α;HER2为人表皮生长因子受体2;CD20为分化簇20;VEGF为血管内皮生长因子。非格司亭(Neupogen®)美国安进(Amgen)公司生产;EP2006(Zarzio®)意大利山德士(Sandoz)公司生产;阿达木单抗(Humira®)美国艾伯维(AbbVie)有限公司生产,国药准字S20191006;MSB11022德国默克(Merck)集团生产;BI 695501(Cyltezo®)德国勃林格殷格翰(Boehringer Ingelheim)公司生产;ABP501(Amjevita®)美国安进(Amgen)公司生产;英夫利昔单抗(Remicade®)荷兰杨森(Janssen)生物制剂公司生产,国药准字S20120012;CT-P13(Inflectra®/Remsima®)韩国赛尔群(Celltrion)公司生产;依那西普(Enbrel®)美国辉瑞(Pfizer)制药有限公司生产,国药准字S20170049;GP2015山德士(Sandoz)公司生产;曲妥珠单抗(Herceptin®)瑞士罗氏(Roche)制药有限公司生产;ABP 980(KanjintiTM)美国安进(Amgen)公司生产;SB3韩国三星Bioepis公司生产;CT-P6韩国赛尔群(Celltrion)公司;利妥昔单抗(MabThera®)瑞士罗氏(Roche)制药有限公司生产,国药准字J20170034、J20170005;CT-P10(Truxima®)韩国赛尔群(Celltrion)公司生产;贝伐珠单抗(Avastin®)瑞士罗氏(Roche)制药有限公司生产,S20170035、S20170036;BEVZ92西班牙Mabxience公司生产
自2003年开始,我国就建立了药品不良反应(ADR)在线自动报告监测系统。除了被动的ADR报告制度外,我国也已开始建立积极的药物警戒监测项目,建立了基于电子健康档案的集中安全监测计划和国家药品不良反应监测前哨联盟计划,进一步加强药品不良反应的报告、监测和分析。但在实践中仍存在一定差距,需在制度建设和完善法律法规方面做出更多努力,以加强药物的安全使用[29]。
此外,生物制剂作为大分子药物存在免疫原性的潜在风险,可以直接诱导机体产生抗药抗体(ADA),会对药物的安全性和有效性产生重大影响。而生物药的免疫原性监测,需要提供足够长的观察周期,以充分暴露药物的潜在风险,但生物类似物在上市前针对免疫原性的评估往往是不充分的[30]。2020年NMPA药审中心颁布的《贝伐珠单抗注射液生物类似药临床试验指导原则(征求意见稿)》中建议在完成主要疗效终点观察后至少收集单药维持治疗1年的安全性数据,生存随访2年数据[31]。
2019年,国家卫生健康委《关于开展药品使用监测和临床综合评价工作的通知》要求充分认识药品使用监测和临床综合评价的重要性,并进一步强调了要围绕药品的安全性、有效性、经济性、创新性、适宜性、可及性进行全面的评价,对生物类似药在临床应用过程的评价方法学研究具有十分重要的指导意义[32]。2020年,我国六部委联合颁布的《关于印发加强医疗机构药事管理促进合理用药的意见的通知》进一步强调要建立健全药品使用监测与临床综合评价工作机制和标准规范,突出药品的临床价值[33]。
生物类似药作为一种上市后需要严密监测的药物之一,应鼓励临床医师、药师和患者报告任何疑似的药物不良反应。目前,我国生物类似药的命名采用通用名[34],无法迅速准确识别药物信息。欧盟生物类似药指南推荐对新上市的或长期使用数据有限的药物,在说明书和包装上会添加黑色倒三角("▼")的警示标志,以提醒用药相关人员加强对该药的额外监测[13]。上述经验可适用于我国生物类似药的安全性监测,值得我们借鉴和学习。
共识一:建议生物类似药在临床应用中,除进行常规安全性监测外,应积极开展RWS和临床综合评价,以指导临床合理用药。(A)
从原则上讲,在满足科学条件的前提下,将生物类似药的适应证从经临床确证的适应证外推(extrapolation of indications)到参照药的其他已批准适应证,是对生物类似药特有的鼓励政策。但必须指出,适应证外推必须要有充分的科学证据支持,应针对每一个药物独立进行评估[35]。世界卫生组织(WHO)规定,生物类似药的适应证不能自动外推至参照药的所有已批适应证。在满足以下所有要求时,方可对适应证进行外推:(1)临床研究采用的是一个可分辨出生物类似药和参照药潜在差异的敏感模型;(2)不同适应证的药物作用机制具有临床相关性和(或)作用受体相同;(3)已经对生物类似药的安全性和免疫原性进行了充分研究,在扩展到其他适应证时不会出现预期的特殊或额外的安全性问题[36,37]。
美国食品药品管理局(FDA)对于适应证外推,同样需要提供充分的科学论证来外推临床数据(表3),以支持每一种使用条件下的生物相似性[38]。

FDA适应证外推摘要[38]
FDA适应证外推摘要[38]
| 监管考虑 | 科学理由 |
|---|---|
| 与参照药适应证 | 外推的适应证须为参照药在本国批准上市的适应证 |
| 外推的适应证与参照药无临床意义的差异 | |
| 使用情况下具有相同的作用机制 | 产品的每个相关活动/功能的靶点/受体与靶点/受体分子的结合,剂量/浓度反应和信号传导模式 |
| 产品结构与靶/受体相互作用之间的关系 | |
| 靶/受体的位置和表达 | |
| 药代动力学相似 | 不同人群中的药代动力学相似 |
| 免疫原性 | 不同人群中的免疫原性相似 |
| 预期毒性 | 不同人群中预期毒性相似(包括预期的毒性是否与产品的药理活性或脱靶效应有关) |
| 可能影响安全性或疗效的其他因素 | 例如:给药途径 |
注:FDA为美国食品药品管理局
EMA认为外推的合理性取决于利用生物相似性研究的完整证据链,包括药学分析、非临床、临床PK/PD和临床疗效/安全性等数据[39]。
2015年我国颁布的《指导原则》中对"适应证外推"是这样描述的:(1)比对试验研究证实临床相似的,可以考虑外推至参照药的其他适应证;(2)对外推的适应证,应当是临床相关的病理机制和(或)有关受体相同,且作用机制以及靶点相同的;临床比对试验中,选择了合适的适应证,并对外推适应证的安全性和免疫原性进行了充分的评估;(3)适应证外推需根据产品特点个案化考虑,对合并用药人群、不同合并疾病人群及存在不同推荐剂量等情形进行适应证外推时应慎重[1]。
同时,生物药由于在国内外获批的适应证多存在差异,在处方审核中应当予以关注。以贝伐珠单抗为例,截至目前其在美国FDA获批的适应证为:晚期非鳞状非小细胞肺癌、转移性结直肠癌、复发性胶质母细胞瘤、转移性肾癌、宫颈癌和卵巢癌等肿瘤适应证[40];而在我国NMPA批准的适应证为转移性结直肠癌,后增加非鳞状非小细胞肺癌的适应证[41]。而生物类似药作为原研生物制剂的类似物,从保证患者安全性与有效性的角度考虑,外推的适应证应是原研药在我国批准的适应证,对于原研药没有在我国获批的适应证,因缺乏我国临床试验和临床使用数据,意味着缺少我国人群的安全性与有效性数据,直接外推使用将带来未知的风险。
2015年,中国药理学会颁布的《超说明书专家共识》对于尚未获得国家监管部门批准的生物类似药的外推使用,需通过医疗机构药事管理与药物治疗委员会和伦理委员会的审批并备案[42]。药事管理与药物治疗委员会审批时,应当组织相关领域专家认真讨论和研究适应证外推的必要性和相关依据,并作出慎重决定,不建议外推为辅助性用药。2020年颁布的《关于加强医疗机构药事管理促进合理用药的意见》强化了药品的合理使用,包括加强医疗机构药品安全管理、提高医师临床合理用药水平、强化药师或其他药学技术人员对处方的审核、加强合理用药管理和绩效考核等4方面的措施[33]。
随着更多生物类似药的研发上市,加强临床医师、药师以及患者对生物类似药,特别是关于适应证外推用药的教育显得尤为重要。目前,德国已将有无积极鼓励针对医生的生物类似药的教育作为药物市场准入政策的一个重要考量方面[43]。医师和药师应当明确说明书中哪些适应证为外推适应证,对其存在的潜在风险保持警惕,并加强用药监测;患者对生物类似药的外推使用应具有知情同意权,对用药过程中疑似不良反应的不适症状应积极主动地向医生或药师报告。多方配合,共同促进生物类似药的临床合理使用。
共识二:如果生物类似药在临床应用中必须外推适应证使用,且尚未获得国家监管部门的批准,建议必须通过医疗机构药事管理与药物治疗委员会审批;对于国家监管部门批准的外推适应证,应当在临床使用过程中加强用药监测。(A)
生物类似药的研发成本相对原研药低,具有一定的价格优势[44],对缓解医疗服务付费方(包括政府、机构与个人)所面临的资金压力,增加药物的可及性,为医生和患者提供更多的治疗选择,具有积极的意义。
据2016年艾美仕(IMS)市场研究报告预测,2016—2020的5年间,在欧洲5国(法国、德国、意大利、西班牙、英国)和美国使用生物类似药后,可累计节省490亿欧元(生物类似药价格为原研药价格的80%)至980亿欧元(生物类似价格为原研药价格的60%)的医疗成本[45]。生物类似药在节省成本方面的优势,不仅仅源于价格因素,而是综合考虑多方面的因素,例如在2013年,EMA批准的皮下注射的曲妥珠单抗类似药非劣效于静脉使用的原研药,但注射的时间更短(5 min比30~90 min),因此是较原研药治疗成本更低的替代药物[46]。例如2019年我国上市的首款生物类似药——利妥昔单抗类似药(HLX01),相较于原研药有价格优势[47],有助于提高生物药的可及性,更好地满足患者对生物治疗药品的需求。
目前发达国家已将生物类似药的药物经济学评估作为药物市场准入与定价政策的重要参考依据[43]。而我国很少开展生物类似药在药物经济学方面的研究,更是缺乏长期疗效与经济性的真实世界证据。2020年颁布的《基本医疗保险用药管理暂行办法(征求意见稿)》中明确了《基本医疗保险药品目录》中纳入和调出的药品提交药物经济学资料的重要性[48]。因此鼓励生物类似药积极开展药物经济学评估,为临床应用和医保决策,提供科学的数据支持。
目前国外在生物类似药的药物经济学研究方面已开展了不少相关研究(表4),对节约医药卫生资源,促进类似药在本国(地区)的良性发展,扩大获益人群具有积极作用,这方面的经验值得我们借鉴学习。中国药学会药物经济学专业委员会颁布的《中国药物经济学评价指南2019》规范了我国在药物经济学方面研究[49],同样适用于生物类似药。由于生物类似药多集中在肿瘤、免疫等领域,患者基本情况和治疗方案均较为复杂,应当结合临床应用实践和治疗目标,设立适宜对照方案和研究角度,确定并收集患者的长期治疗结局指标,主动进行生命质量指标调查并将之纳入治疗结局指标,同时收集治疗费用数据,结合模型方法开展生物类似药的长期成本-效益研究,为临床和医保决策提供参考。
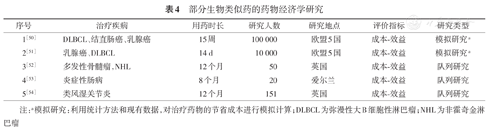
部分生物类似药的药物经济学研究
共识三:建议在临床应用中积极开展基于真实世界数据的药物经济学研究;药物经济学研究中应综合考虑患者长期治疗结局、生命质量改善以及长期总费用。(A)
2018年颁布的《医疗机构处方审核规范》中对西药及中成药处方审核的项目做出了明确规定,这些规定涵盖了临床常见的药物审核需求[55]。同年颁布的《关于加快药学服务高质量发展的意见》再次强调了加强处方审核和处方点评的要求[56]。在临床实际使用过程中,对于超适应证使用的情况,需以患者获益和知情为前提,并有合理的医学实践证据,且经医院药事管理与药物治疗学委员会的批准[57]。在临床实际使用中,存在现超适应证使用情况(见本《共识》"生物类似药的适应证管理"章节)。同时,联合治疗是针对肿瘤治疗耐药的基本策略[58],抗肿瘤单抗在临床中大量存在与传统化疗药、小分子化学靶向药、或其他生物制剂联合使用的情况。如帕妥珠单抗联合曲妥珠单抗和多西他赛治疗人表皮生长因子受体2阳性的转移乳腺癌能让患者显著获益[59]。而反观生物类似药所开展的与参照药的等效研究是曲妥珠单药联合化疗的头对头比较[60]。除此之外,生物类似药的超剂量用药也是需要重点关注的问题。以上这些情况都对生物类似药在临床上的应用提出了挑战,因此药师在做好生物类似药常规处方审核的同时,需加强对这些方面的关注。
2019年,中国抗癌协会肿瘤临床药学专业委员会与中国药师会肿瘤药师分会共同组织全国的药学专家制定了《抗肿瘤药物处方审核专家共识——肺癌》[61]、《抗肿瘤药物处方审核专家共识——结直肠癌》、《抗肿瘤药物处方审核专家共识——肝癌》[62]3个专家共识,一致提出运用"六步法"进行抗肿瘤药物处方审核:即合法性审核→患者评估审核→方案审核→器官功能及实验室指标审核→预处理审核→非常规处方复核。同时对包括:患者特殊(即指:属于抗肿瘤药物禁忌使用人群的患者)与治疗方案特殊(即指:非指南共识推荐方案;非备案的药品超说明书使用,包括剂量、浓度、给药途径、给药顺序、适应证等)的"非常规处方复核",建议提请上级药师或多学科团队进行再次审核。
共识四:建议药师加强生物类似药在超剂量用药、超适用人群等方面的处方审核和点评。(A)
生物类似药在我国的发展已进入快速发展期。虽然我国在生物类似药临床应用管理的指导规范与实践经验上与欧美发达国家相比尚存差距,但随着更多生物类似药上市,生物类似药必将在临床上发挥更重要的作用。而本《共识》的制定旨在规范生物类似药的临床应用管理,推动临床医生和药师共同促进生物类似物在中国的良性发展,并在进一步实施过程中不断积累经验,让更多患者受益。
编写组长
童荣生(四川省医学科学院四川省人民医院);赵杰(郑州大学第一附属医院)
共识制定组成员(按姓氏拼音排序)
巴桑拉姆(西藏自治区人民医院);曹力(南昌大学第一附属医院);陈万生(海军军医大学附属长征医院);陈孝(中山大学附属第一医院);陈耀龙(兰州大学);陈英(广西壮族自治区人民医院);邓建雄(广东省药品不良反应监测中心);董梅(哈尔滨医科大学附属肿瘤医院);杜光(华中科技大学同济医学院附属同济医院);杜智敏(哈尔滨医科大学附属第二医院);方晴霞(浙江省人民医院);封卫毅(西安交通大学第一附属医院);高申(海军军医大学附属长海医院);葛卫红(南京大学医学院附属鼓楼医院);龚志成(中南大学湘雅医院);郭代红(中国人民解放军总医院);郭瑞臣(山东大学齐鲁医院);郭玉金(济宁市第一人民医院);侯锐钢(山西医科大学第二医院);胡明(四川大学华西药学院);胡欣(北京医院);黄品芳(福建医科大学附属第一医院);菅凌燕(中国医科大学附属盛京医院);姜玲(安徽省立医院);李焕德(中南大学湘雅二医院);李丽(海南医学院第一附属医院);刘皋林(上海市第一人民医院);刘景丰(福建医科大学附属肿瘤医院);刘丽宏(首都医科大学附属北京朝阳医院);刘世霆(南方医科大学南方医院);刘小玲(内蒙古自治区人民医院);卢海儒(青海省人民医院);吕迁洲(复旦大学附属中山医院);马瑞莲(内蒙古医科大学附属医院);梅丹(中国医学科学院北京协和医院);缪丽燕(苏州大学附属第一医院);齐晓勇(河北省人民医院);宋金春(武汉大学人民医院);隋忠国(青岛大学附属医院);孙洲亮(厦门大学附属第一医院);童荣生(四川省医学科学院四川省人民医院);王家伟(北京同仁医院);王建华(新疆医科大学第一附属医院);文爱东(空军军医大学西京医院);文友民(宁夏医科大学总医院);武新安(兰州大学第一医院);夏培元(陆军军医大学西南医院);胥婕(北京大学第三医院);杨宏昕(内蒙古自治区人民医院);杨婉花(上海交通大学医学院附属瑞金医院);于倩(吉林大学中日联谊医院);张抒扬(中国医学科学院北京协和医院);张玉(华中科技大学同济医学院附属协和医院);张相林(北京中日友好医院);张幸国(浙江大学附属第一医院北伦分院);张晓坚(郑州大学第一附属医院);张健(上海交通大学医学院附属新华医院);张伶俐(四川大学华西第二医院);张鉴(山东省立医院);张抗怀(西安交通大学第二附属医院);张伟(河南省人民医院);张永军(石河子大学医学院第一附属医院);张志清(河北医科大学第二医院);张志仁(哈尔滨医科大学附属肿瘤医院);赵杰(郑州大学第一附属医院);赵荣生(北京大学第三医院);赵志刚(首都医科大学附属北京天坛医院);赵庆春(中国人民解放军北部战区总医院);郑志昌(贵州医科大学附属医院);左笑丛(中南大学湘雅三医院)
咨询专家组成员(按姓氏拼音排序)
巴一(天津医科大学肿瘤医院);陈新石(中华医学杂志编辑部);韩冰(中国医学科学院北京协和医院);李卉(四川省肿瘤医院);梁寒(天津医科大学肿瘤医院);林琳(中国医学科学院肿瘤医院);刘继红(中山大学附属肿瘤医院);刘云鹏(中国医科大学附属第一医院);刘真真(河南省肿瘤医院);马军(哈尔滨血液肿瘤研究所);欧阳取长(湖南省肿瘤医院);潘宏铭(浙江大学医学院附属邵逸夫医院);潘志忠(中山大学附属肿瘤医院);沈志祥(上海交通大学医学院附属瑞金医院);宋聚良(黑龙江省牡丹江林业中心医院);束永前(江苏省人民医院);佟仲生(天津医科大学肿瘤医院);王风华(中山大学附属肿瘤医院);王晓稼(中国科学院大学附属肿瘤医院浙江省肿瘤医院);吴德沛(苏州大学附属第一医院);吴凤英(上海市肺科医院);徐兵河(中国医学科学院肿瘤医院);徐惠绵(中国医科大学附属第一医院);尹如铁(四川大学华西第二院);张清媛(黑龙江省肿瘤医院);赵群(河北省肿瘤医院);周蕾蕾(四川省医学科学院四川省人民医院);周琦(重庆大学附属肿瘤医院);朱军(北京大学肿瘤医院)
所有编写组成员均声明不存在利益冲突
























