
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
脊髓性肌萎缩症(spinal muscular atrophy,SMA)是儿童最常见的神经肌肉病,以脊髓前角α-运动神经元退化变性导致的肌无力和肌萎缩为主要临床特征。本共识中SMA特指位于5q13的运动神经元存活基因1(SMN1;OMIM 600354)致病性变异所导致的5q-SMA。SMA发病率约为1/10 000,人群携带率约为1/50[1]。2019年中国大陆上市了疾病修正治疗药物诺西那生钠注射液,也相继发表了SMA多学科管理专家共识[2],标志着SMA在我国进入了一个全新的精准诊治和管理时期。SMA的携带者和新生儿筛查在一些国家和地区已常规开展[3,4],我国一些地区也逐渐开始筛查[5,6],SMA预防窗口进一步提前。
SMA的致病基因SMN1和修饰基因SMN2(OMIM 601627)高度同源,SMN1决定疾病的发生,SMN2影响疾病的严重程度和进展,使得SMA的遗传学诊断不同于绝大多数单基因遗传病。规范SMA遗传学诊断及应用对于临床诊治、管理、预防和遗传咨询将提供重要帮助。本共识参照国内外近年SMA临床诊疗实践和指南共识[2,7,8,9,10],由具有实践经验的多学科专家研究起草,包括了患者和携带者基因型、基因诊断技术的适用性和局限性,以及基因诊断、产前诊断、植入前遗传学检测和携带者筛查的要点及遗传咨询等内容,并对SMN2拷贝数的临床价值提出了一些建议。旨在为医生和实验室人员的临床实践提供指导帮助。
SMA患者起病年龄差异性大,从出生前至成人期均可发病。主要表现为以四肢近端为主的进行性肌无力和肌萎缩,随着疾病进展,可出现呼吸、消化、骨骼等多系统受累。根据起病年龄、运动里程碑及病情进展程度,SMA分为五型。近年的临床实践趋于将每型SMA进一步分为亚型,以便更好地理解自然病程和观察药物疗效(表1)[11,12,13]。

脊髓性肌萎缩症的分型和临床表现
脊髓性肌萎缩症的分型和临床表现
| 型别 | OMIM | 起病年龄 | 运动里程碑 | 临床表现 | 自然病程 | 比例 | SMN2拷贝数 |
|---|---|---|---|---|---|---|---|
| 0 | - | 出生前/出生时 | 无 | 除眼球外,几乎无肢体、躯干和面部的任何活动,无吸吮动作;先天性关节挛缩、肌肉萎缩、反射消失;出生后即需要呼吸机辅助呼吸;可合并先天性心脏病 | 数月 | 很少 | 1 |
| 1 | 253300 | <6个月 | 不能独坐 | 松软儿,严重肌张力低下,四肢无力;舌肌、面肌、咀嚼肌无力;胸廓钟型;容易反复呼吸道感染及呼吸衰竭 | 1a和1b≤2岁,1c的2岁生存概率为95% | 40%~50% | 1a主要为1;1b主要为2;1c主要为3 |
| 2 | 253550 | 6~18个月 | 能独坐,不能独立行走 | 婴幼儿期出现缓慢加重的全身性肌无力和肌张力低下;运动发育落后,舌肌纤颤或手部肌束颤;可伴关节挛缩及脊柱侧弯,影响呼吸功能 | 大部分可生存至成年 | 30%~40% | 主要为3 |
| 3 | 253400 | >18个月~10岁 | 能独立行走 | 生后1年内运动发育正常;儿童期逐渐出现近端为主的肌无力,下肢重于上肢;疾病进展丧失行走能力;可见肌束颤;后期出现脊柱侧弯、关节畸形、呼吸功能不全等表现 | 寿命不缩短或轻度下降 | 10%~20% | 3或4 |
| 4 | 271150 | 成人期 | 能跑跳等所有运动能力 | 青少年期或成人期起病,下肢起始的四肢近端无力,病情缓慢进展 | 寿命一般不受影响 | 较少 | 主要为4 |
注:在1型亚分型中,1a型在出生后前两周内发病,无任何运动功能;1b型为3个月前发病,控头困难,不能翻身;1c为3~6个月发病,可以控头和翻身;"-"为无相关内容
(1)临床评估:临床医师根据病史查体拟诊,主要临床特点为进行性、对称性四肢和躯干的肌无力,近端重于远端,下肢重于上肢,有时可见舌肌纤颤、手震颤;(2)临床检测:包括血肌酶谱,肌酸激酶(CK)值正常或轻度升高,绝大多数患者不超过正常值的10倍,肌电图提示神经源性损害;(3)基因检测显示SMN1外显子7纯合缺失或SMN1复合杂合突变,阳性结果可确诊SMA;(4)基因检测阴性结果患者需行肌电图及肌肉活检,帮助诊断与鉴别诊断[11](图1)。SMA的临床分型主要依据患者起病年龄和获得的运动里程碑,并参考SMN2拷贝数(表1)。部分患者的运动里程碑获得迟于健康个体,因此,建议对患者进行定期随访。
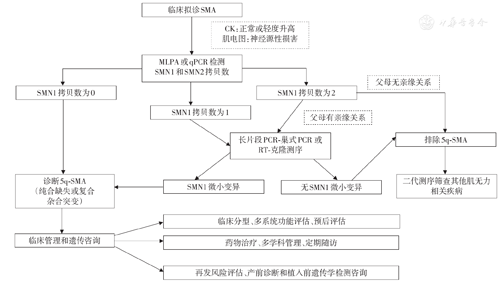
注:SMA为脊髓性肌萎缩症;CK为肌酸激酶;MLPA为多重连接依赖性探针扩增;qPCR为实时荧光定量PCR;RT为逆转录
当疑似患者的基因检测未见SMN1双等位基因致病变异,或症状不典型,或伴有非SMA临床表现时,进行以肌无力为主要症状的其他疾病的鉴别诊断非常必要。6个月以下患儿需鉴别其他软婴综合征,6~18个月患儿需鉴别婴幼儿时期起病的神经肌肉病。成年期患者需鉴别成人期起病的神经肌肉病。因需鉴别的疾病种类繁多,常选择二代测序技术(NGS)及其他高通量诊断技术(表2)。

SMA的鉴别诊断
SMA的鉴别诊断
| 起病年龄与临床分型 | 疾病名称 | 临床鉴别要点 | 致病基因 | 检测方法 |
|---|---|---|---|---|
| <6个月(SMA 0或1型) | 脊髓性肌萎缩症伴呼吸窘迫1型(SMARD1) | 膈肌麻痹,呼吸窘迫明显;肢体远端无力明显。2岁后常病情稳定甚至部分好转 | ICHMBP2 | NGS |
| X连锁婴儿型脊髓性肌萎缩症(SMAX2) | 与SMA-0型表现类似 | UBA1 | NGS | |
| 先天性肌无力综合征 | 眼睑下垂、眼肌麻痹;症状有波动性 | CHRNA1, CHRNB1, CHRND, CHRNE, COLQ, CHAT, SYT2. AGRN, MUSK, DOK7, RAPSN, GFPT1, DPAGT1, ALG2, ALG14, SCN4A, LRP4, SNAP25, COL13A1, SLC5A7, SLC18A3, PREPL, SLC25A1, MYO9A, VAMP1等 | NGS | |
| 先天性肌病 | 亦以近端无力明显;肌电图、肌肉活检有助于鉴别 | ACTA1, NEB, TPM2, TPM3, TNNT1, CFL2, KTB-DB13, RYR1, SELENON, MTM1, DNM2, BIN1等 | NGS | |
| 先天性肌营养不良 | 亦常合并关节畸形;肌电图、肌肉活检有助于鉴别 | LAMA2, COL6A1, COL6A2, COL6A3, POMT1,POMT2, POMGNT1, FKTN, FKRP, LARGE,ISPD, GTDC2, DAG1, TMEM5, B3GALNT2,SGK196, B3GNT1, GMPPB等 | NGS | |
| 先天性强直性肌营养不良 | 特殊面容 | DMPK | TP-PCR | |
| 糖原累积病Ⅱ型 | 肥厚性心肌病,舌体肥厚,肝脾大;血酸性α-葡萄糖苷酶活性降低 | GAA | NGS或Sanger测序 | |
| Prader-Willi综合征 | 生殖系统发育异常;肌张力缓慢恢复 | 父源15q11微缺失 | MS-PCR、MS-ML - PA、CMA | |
| Zellweger综合征 | 头面部畸形、视听觉障碍、癫痫、肝大 | PEX1, PEX5,PEX12,PEX6,PEX2,PEX10,PEX26, PEX16, PEX3, PEX13, PEX19, PEX14等 | NGS | |
| 其他获得性疾病:缺血缺氧性脑脊髓病、高颈段脊髓损伤 | 仔细询问病史予以鉴别 | - | - | |
| 6~18个月(SMA 2或3型) | Duchenne/Becker型肌营养不良 | 双腓肠肌假肥大;肌酶显著升高;肌电图、肌肉活检有助于鉴别 | DMD | MLPA, NGS |
| 肢带型肌营养不良(LG-MD) | 肌酶明显升高,肌电图、肌肉活检有助于鉴别 | CAPN3, DYSF, SGCG, SGCA, SGCB, SGCD, TCAP, TRIM32, TTN, ANO5, PLEC, TRAP-PC11, POGLUT1, DNAJB6, TNP03, HNRNP-DL, TFG等 | NGS | |
| 非5q脊髓性肌萎缩症 | 病程进展常更缓慢 | VAPB, CHCHD10, TRPV4, DYNC1H1, BICD2等 | NGS | |
| 轴索性遗传性运动神经病或运动感觉周围神经病 | 肢体远端无力萎缩明显;肌电图有助于鉴别 | SIGMAR1, PLEKHG5, DNAJB2, HSPB8, HSPB1, HSPB3,GARS,BSCL2,DCTN1等 | NGS | |
| 脊髓性肌萎缩症叠加疾病 | 合并严重关节挛缩、小脑萎缩、肌阵挛癫痫等其他表现 | GLE1, VRK1, EXOSC3, SLC52A3, SLC52A2,ASAH1, ATP7A等 | NGS | |
| 晚发型神经节苷脂贮积症 | 同时累及大脑、小脑,出现智能减退、共济失调、肌张力障碍等表现。血酶学检测可诊断 | HEXA | NGS或Sanger测序 | |
| 其他疾病:重症肌无力、格林巴利综合征 | 起病形式、肌电图、腰穿脑脊液检查等予以鉴别 | - | - | |
| 成人期(SMA4型) | 脊髓延髓性肌萎缩症(Kennedy病) | 男性患病,中年起病,可伴随雄激素功能不足其他表现;肌电图有助于鉴别 | AR | Sanger测序 |
| 家族性肌萎缩侧索硬化 | 病情进展更快,伴随上运动神经元损害 | SOD1, FUS, VAPB, ANG, TARDBP, FIG4, OPTN, VCP, UBQLN2, CHMP2B, PFN1, ERBB4, HNRNPA1, MATR3, TUBA4A, ANXA11, C9ORF72 | NGS或长读长三代测序 |
注:此表参考GeneReviews(https://www.ncbi.nlm.nih.gov/books/NBK1352/)和UpToDate(www.uptodate.com/contents/search)网站;SMA为脊髓性肌萎缩症;NGS为二代测序技术;TP-PCR为三引物PCR法;MS-PCR为甲基化特异性PCR;MS-MLPA为甲基化特异性多重连接探针扩增;CMA为染色体微阵列分析;MLPA为多重连接探针扩增;"-"为无相关内容
SMA为常染色体隐性遗传病。致病基因SMN1(NG_008691.1)定位于5q13.2,转录本编号NM_000344.3,其编码的全长SMN蛋白(NP_000335.1)在各组织细胞广泛表达,参与剪接体蛋白复合体的组装,是真核细胞生物生存所必需的管家蛋白[14]。SMN1双等位基因发生致病性变异通常导致SMA发生。SMA病理生理学表现为脊髓前角α-运动神经元退行性病变和神经肌肉接头发育异常。
98%的SMA患者的SMN1双等位基因变异分别遗传自双亲,2%患者有一个等位基因发生新生变异[15]。SMA突变基因型主要有两类:95%由SMN1双等位基因纯合缺失所致,即[0+0]基因型;5%由SMN1复合杂合突变所致,即一个等位基因缺失,另一个等位基因发生微小致病性变异,为[0+1d]基因型。SMN1双等位基因均为微小致病性变异,即[1d+1d]基因型,非常罕见,目前仅有白种人近亲婚配的病例报道。SMN1缺失大部分为外显子7合并外显子8共同缺失,少部分仅为外显子7缺失。由于外显子8位于非编码区,SMN1缺失通常指外显子7缺失。SMN1微小致病性变异在不同种族患者中表现不同的变异谱系,目前国内外已报道的微小致病性变异约90种(http://www.hgmd.cf.ac.uk),中国患者已报道[16,17,18,19,20,21,22]近30种,其中检测频次≥2,并经美国医学遗传与基因组学学会(ACMG)致病性评估[23,24]确认为是致病性微小变异有7种(表3)。
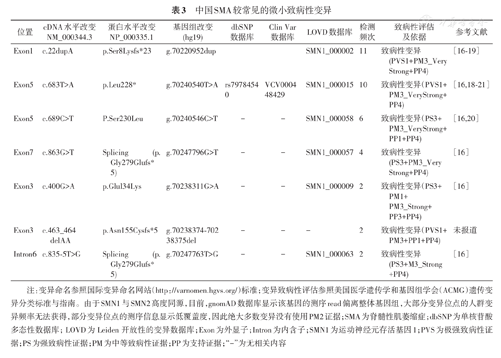
中国SMA较常见的微小致病性变异
中国SMA较常见的微小致病性变异
| 位置 | cDNA水平改变NM_000344.3 | 蛋白水平改变NP_000335.1 | 基因组改变(hg19) | dbSNP数据库 | Clin Var数据库 | LOVD数据库 | 检测频次 | 致病性评估及依据 | 参考文献 |
|---|---|---|---|---|---|---|---|---|---|
| Exon1 | c.22dupA | p.Ser8Lysfs*23 | g.70220952dup | SMN1_000002 | 11 | 致病性变异(PVS1+PM3_Very Strong+PP4) | [16,17,18,19] | ||
| Exon5 | c.683T>A | p.Leu228* | g.70240540T>A | rs7978454 0 | VCV0004 48429 | SMN1_000015 | 10 | 致病性变异(PVS1+PM3_VeryStrong+PP4) | [16,18,19,20,21] |
| Exon5 | c.689C>T | P.Ser230Leu | g.70240546C>T | - | - | SMN1_000058 | 6 | 致病性变异(PS3+PM3_VeryStrong+PP1+PP4) | [16,20] |
| Exon7 | c.863G>T | Splicing(p.Gly279Glufs*5) | g.70247796G>T | - | - | SMN1_000057 | 4 | 致病性变异(PS3+PM3_Very Strong+PP4) | [16] |
| Exon3 | c.400G>A | p.Glul34Lys | g.70238311G>A | - | - | SMN1_000009 | 2 | 致病性变异(PS3+PM1+PM3_Strong+PP3+PP4) | [16] |
| Exon3 | c.463_464 delAA | p.Asn155Cysfs*5 | g.70238374-70238375del | - | - | - | 2 | 致病性变异(PVS1+PM3+PP1+PP4) | 未报道 |
| Intron6 | c.835-5T>G | Splicing(p.Gly279Glufs*5) | g.70247763T>G | - | - | SMN1_000063 | 2 | 致病性变异(PS3+M3_Strong+PP4) | [16] |
注:变异命名参照国际变异命名网站(http://varnomen.hgvs.org/)标准;变异致病性评估参照美国医学遗传学和基因组学会(ACMG)遗传变异分类标准与指南。由于SMN1与SMN2高度同源,目前,gnomAD数据库显示该基因的测序read偏离整体基因组,大部分变异位点的人群变异频率无法获得,部分变异位点的测序信息显示低覆盖度,因此绝大多数变异没有使用PM2证据;SMA为脊髓性肌萎缩症;dbSNP为单核苷酸多态性数据库;LOVD为Leiden开放性的变异数据库;Exon为外显子;Intron为内含子;SMN1为运动神经元存活基因1;PVS为极强致病性证据;PS为强致病性证据;PM为中等致病性证据;PP为支持证据;"-"为无相关内容
SMN2全长mRNA编码与SMN1相同的SMN蛋白。SMN2与SMN1的序列同源性>99.9%,两者仅存在5个碱基差异,其中在第7外显子第6位c.840的C/T,导致90%的SMN2 mRNA外显子7被选择性剪接,仅有10%的SMN2表达全长有功能SMN蛋白[25]。SMN1和SMN2均位于5q13.2,该区域存在包括SMN在内的多对同源基因导致基因组不均等的互换和基因转换,出现SMN1和SMN2拷贝数的多种变化。在SMA患者中可以见到SMN1真实缺失,SMN1∶SMN2拷贝数通常为0∶2,约占中国SMA人群28%[16];还有SMN1转换为SMN2导致的SMN1缺失和SMN2增加,或者SMN1真实缺失伴随SMN2重复,这两类情况SMN1∶SMN2拷贝数为0∶3或者0∶4,约占62%[16];同时,SMN1和SMN2之间还存在部分转换,出现SMN1-SMN2融合基因,患者仅见SMN1外显子7缺失,外显子8不缺失,约占6%[16]。在一般人群中正常个体的SMN1基因至少为1拷贝,而SMN2基因可以为0拷贝,绝大数个体的SMN1∶SMN2拷贝数为2∶2,但也有正常个体SMN1基因多于2拷贝。
SMN2拷贝数是目前公认的SMA修饰因子,患者携带SMN2拷贝数越多表型越轻,尽管其与表型的相关性不完全一致,在国内外管理共识中仍将SMN2拷贝数作为SMA诊断的标准步骤之一[2,7]。一般认为,携带1个SMN2拷贝的患者是最严重的0型,宫内发病,出生后1个月内死亡;携带2个SMN2拷贝的患者通常为1型,大部分在2岁前死亡,早期给予药物治疗及呼吸和营养支持可降低死亡率;携带3个SMN2拷贝的患者主要为2型和3型,需要药物治疗和前瞻性干预及定期随访;携带4个SMN2拷贝数的患者通常为4型,发病晚,病情进展缓慢[26,27]。SMN2拷贝数在以调控SMN蛋白为治疗策略的药物临床试验中常常被作为重要参考数据[28],而在新生儿筛查中则被作为症状出现前患者治疗评估的重要生物学标志物[9]。
SMA携带者指表型正常但一条染色体上的SMN1基因功能正常而另一条染色体的SMN1基因存在致病性变异的个体。在正常等位基因中,将携带1个SMN1基因拷贝定义为1拷贝(1-copy);将携带2个SMN1基因拷贝定义为2拷贝(2-copy)。在致病等位基因中,将携带SMN1缺失或因转换导致的SMN1缺失定义为0拷贝(0-copy);将携带微小变异的SMN1基因定义为1d[8]。因此,SMA携带者的SMN1基因型主要分为4种:[1+0]基因型,即外显子7拷贝数为1,该携带者的一条染色体上SMN1功能正常,另一条染色体上存在SMN1缺失或转换;[2+0]基因型,即SMN1外显子7拷贝数为2,该携带者2个功能正常的SMN1基因拷贝位于同一条染色体,另一条染色体上存在SMN1缺失或转换;[1+1d]和[2+1d]基因型,即SMN1外显子7拷贝数≥2,一条染色体上的SMN1功能正常,并且拷贝数为1或2,另一条染色体上的SMN1基因存在微小变异而功能异常[8]。世界范围内SMA携带率为1/100~1/45[1],亚洲人群携带率约为1/48。一般人群中,[1+0]、[2+0]、[1+1d]和[2+1d]基因型的人群频率分别为2.58×10-2、1.09×10-3、4.72×10-4和1.99×10-5[29]。而在肯定携带者人群中[2+0]频率约为4%[30],[1+1d]频率约为5%[16]。
SMN1发生双等位基因的致病性变异是SMA诊断的主要依据,临床诊断或临床疑似SMA的患者均应进行基因检测确诊。检测的目标基因为SMN1和SMN2。其中SMN1拷贝数和致病性变异的检测结果用于疾病诊断或排除诊断,SMN2拷贝数的检测结果作为患者诊断后的治疗、临床管理和预后评估的参考指标。
SMA基因诊断应满足各种SMN1突变基因型患者的诊断,包括SMN1纯合缺失患者,即[0+0]基因型;以及SMN1基因复合杂合突变患者,即[0+1d]基因型:(1)在人类基因组中SMN1与SMN2高度同源,基因诊断技术须分别针对SMN1和SMN2。SMN1与SMN2全长转录本在外显子7(c.840C/T)和外显子8(c.*239G/A)的差异碱基位点作为区分两个基因座的主要参考位点。(2)SMN1与SMN2均存在多个转录本,为减少致病性变异的误判,推荐SMN1 NM_000344.3和SMN2 NM_017411.3全长转录本分别作为SMN1与SMN2基因分析的参考序列。(3)应用定量分析技术进行基因检测时,建议同时报告SMN1和SMN2的拷贝数。(4)基因诊断明确的患者,其父母有必要进行SMN1检测,以便确定父母SMN1基因型和患者变异基因的来源,进行遗传咨询。
由于95%的SMA患者为SMN1外显子7纯合缺失,应首先进行SMN1拷贝数定量分析。当受检者为SMN1外显子7或外显子7与8纯合缺失,即可诊断为SMN1纯合缺失型患者。当受检者为SMN1杂合缺失,提示需进行另一个SMN1等位基因的序列分析:如检测到微小致病性变异,该个体诊断为SMN1缺失和微小变异的复合杂合突变患者;如未查到微小致病性变异且临床表现又不典型,该个体可能仅为SMN1缺失携带者,可能是其他疾病患者,需进一步鉴别诊断。当受检者没有检测出SMN1纯合缺失或复合杂合变异,但具有明确的SMA临床诊断,则可能存在检测范围外的致病性变异,特别当患者父母为近亲婚配时,应进行SMN1基因全序列分析,以确诊SMN1双等位基因均为致病性微小变异的患者。基因诊断流程见图1。
MLPA方法针对SMN1与SMN2基因第7和第8外显子碱基差异位点(c.840位点C/T和c.*239位点G/A)设计杂交探针,采用其他染色体位点的多个管家基因作为内参基因,以SMN1∶SMN2不同拷贝数的样本作为平行对照,在完成杂交连接等系列反应后,根据荧光峰面积的比值比来判断目标基因序列的拷贝数。MLPA技术通过直接检测SMN1拷贝数,明确区分患者、携带者及正常人;还可同时检测患者的SMN2拷贝数,是目前国内外SMA管理共识推荐使用诊断SMA的金标准[2,7]。但是该方法不能检测SMN1基因微小变异与SMN1[2+0]基因型。
主要原理是通过多重实时荧光定量PCR反应,以管家基因序列为内参,采用Taqman探针法分别对SMN1基因第7和第8外显子进行拷贝数相对定量,来判断是否发生缺失变异。该方法优势是操作简便、成本低廉,适用于人群筛查,但其特异性相比MLPA方法略逊,且SMN2拷贝数需要另外设计探针进行检测。同样不能检测SMN1基因微小变异和[2+0]基因型。
SMN1复合杂合突变患者需要确认发生在SMN1基因的微小变异。由于SMN1和SMN2高度同源,通常采用SMN1特异性长片段PCR结合巢式PCR的方法或SMN1基因逆转录(RT)-克隆测序进行SMN1的变异分析。
根据SMN1和SMN2差异碱基,设计等位基因特异性引物用以特异性扩增整个SMN1来源的基因组DNA序列,再以长片段扩增产物作为模板通过巢式PCR扩增每个外显子及邻近的内含子进行序列分析,检测SMN1序列是否存在基因变异。可以检测SMN1外显子和邻近内含子区域的微小变异。
提取患者外周血总RNA,逆转录后非特异性扩增全长SMN基因(含SMN1和SMN2)cDNA,根据SMN1和SMN2差异碱基挑选SMN1克隆,通过Sanger测序的方法检测来源于SMN1 cDNA的基因变异。该方法可以检测外显子的变异,也可以检测内含子区域影响剪接的变异的异常转录本,但是不能检测到调控区域的变异。
因操作简便,可用于筛查SMN1杂合缺失患者是否存在SMN基因微小变异。但因该方法无法区分微小变异发生在SMN1还是SMN2,需要进一步验证。
由于高通量且商业化,NGS已成为单基因遗传病基因诊断的首选技术。由于SMN1和SMN2基因高度同源性,并且存在因相互部分转换形成的SMN1-SMN2融合基因,而目前的测序技术读长短(比如Illumina测序平台读长为100-150bp),因此NGS数据的常规比对拼接方法无法区别SMN1和SMN2基因的变异,gnomAD数据库内关于SMN1基因变异位点的信息描述也说明这个事实。但由于SMN1和SMN2基因存在2个碱基差异位点(exon7的c.840C/T;exon8的c.*239G/A),实验室可利用该位点的碱基类型的read比例来推测SMN1基因是否存在杂合或纯合缺失,但无法确定SMN1基因缺失长度和SMN2基因拷贝数。2017年一项研究证实通过改进实验操作和生物信息学分析流程[31],NGS可同时检测SMN1基因拷贝数和筛查SMN基因微小变异。与MLPA技术相比,其检测SMN1基因拷贝数变异的灵敏度可达100%,特异性可达99.6%,但应用该技术筛选出的微小变异未能直接明确存在于SMN1或SMN2基因。2020年,另一项研究也建立了用于筛查SMN1和SMN2拷贝数的NGS,应用已知阳性病例通过MLPA技术验证,该技术准确率达到100%[32]。值得注意的是,以上两项研究均将SMN1和SMN2基因(chr5∶70220768-70248838和chr5∶69345350-69373418)都作为靶向区域筛选变异,基于SMN1和SMN2基因的差异性位点对常规测序和生物信息学流程进行改进,使用已知阳性样本的NGS数据作为内标构建SMN1基因的拷贝数计算公式。以上研究表明,NGS直接计算SMN1拷贝数的技术将逐渐成熟和准确,但筛查或诊断SMN1微小变异仍存在很大困难。
目前在我国,NGS尚未成为SMA的常规检测方法,但是其适用于SMA鉴别诊断,即对非5qSMA的神经肌肉病,或者以肌无力为临床症状需要排除诊断的患者开展致病基因变异筛查。如果诊断实验室只是利用SMN1和SMN2基因的差异位点来推测SMN1基因的杂合或纯合缺失,而没有如以上文献建立SMN1基因拷贝数变异的计算方法,则必须使用经典拷贝数诊断方法(比如MLPA)进行验证。如果利用NGS筛查SMN1基因的微小变异,则需要说明如何确定该变异发生在SMN1基因。
SMA传统的检测方法还有双侧双重等位基因特异性PCR(AS-PCR)与聚合酶链反应-变性高效液相色谱(PCR-DHPLC)等。对于不具备定量检测技术条件的检测机构可以应用定性检测技术诊断SMN1外显子7纯合缺失病例,例如PCR-酶切分析或者Sanger测序等。但定性检测技术不能检测SMN1基因的杂合缺失,可能导致复合杂合突变患者的漏诊或误诊。
包括被检测者的个体识别信息,如姓名、性别、出生日期、采集样本时间、样本性质、门诊号及住院号、送检医生信息等。
包括患儿症状、体征、辅助检查结果和临床诊断。
应说明使用的检测方法、检测试剂名称和型号、列出检测方法的适用范围、检测技术的局限性。还应说明SMA的基因型分布的背景资料。
检测的基因名称、基因组位置、基因的转录本号。
(1)拷贝数检测报告应包含如下信息:SMN1基因和SMN2基因外显子7和外显子8的拷贝数;(2)基因微小变异分析报告或基因序列分析报告应包含如下信息:SMN1微小变异信息、遗传模式、变异来源、致病性分析等;(3)建议拷贝数和基因微小变异的检测结果报告以表格形式报告,并提供原始的实验结果图。
SMN1基因外显子7拷贝数缺失为明确的致病性变异,单碱基或几个碱基的微小变异建议参照ACMG制定的基因变异解读指南[23],将变异的临床意义依据评估证据进行五级分类:致病性、可能致病性、临床意义不明、可能良性和良性。致病性和可能致病性变异通常认为是导致疾病发生的变异,临床意义不明变异建议结合临床诊断进行后期数据重分析。对于测序发现的未报道、变异,尤其错义变异,由于缺乏SMN1基因区域的正常人群频率,致病性评估中PM2证据可能无法使用。致病性变异命名参照国际变异命名网站(http://varnomen.hgvs.org/)的标准。
检测报告应给出明确的检测结论。如本检测方法不能完成患者基因诊断,则应给出是否需要进一步验证以及验证方法的建议。
首先判读基因检测报告是否明确了患者的基因型及变异的致病性等,检测结果是否可以解释患者的临床表现,确认遗传学诊断的有效性。对患者及时进行临床分型及病情评估,解释疾病的发生发展,开展药物治疗、多学科管理和定期随访。延长患者生存周期,提高运动功能,延缓和减轻并发症的发生发展。提供患者及监护人遗传咨询,包括:遗传病因、传递模式、再发风险评估、产前诊断或植入前遗传学检测等再生育选择以及家庭成员携带者筛查建议等相关信息和指导(图1)。
由于SMA的病情严重、治疗费用昂贵,现阶段产前诊断仍然是SMA主要的预防手段。
SMA的产前诊断应在具备产前诊断资质的医疗机构由具有资质的专业人员进行。实施产前诊断之前必须预分析或验证先证者及父母的变异基因型,明确先证者的SMN1变异类型,据此制定该家系实施产前诊断的策略和诊断技术。产前诊断通常在母孕的早、中期采集胎儿绒毛组织或羊水细胞进行检测。
(1)生育过SMA患儿的夫妻;(2)夫妻双方均为SMA携带者;(3)生育过临床诊断SMA的患儿,但患儿夭折前未行基因诊断,再生育前通过SMN1基因检测明确双方为SMA携带者的夫妻。
SMA产前诊断的检测基因为SMN1。由于SMN1基因的特殊性,大部分产前诊断机构仅针对SMN1缺失型SMA进行产前检测;复合杂合变异的SMA家庭行产前诊断前应到有检测SMN1微小变异经验的实验室进行微小变异的确定,再进行产前检测。对于SMN1缺失型SMA,要参考父母的基因型对胎儿进行基因型判断。当父母皆为[1+0]基因型,胎儿为SMN1纯合缺失型时诊断为SMA受累胎儿。建议进一步检测SMN2的拷贝数,在产前遗传咨询中可以给予胎儿父母更多关于SMA临床表型信息,帮助他们对胎儿去留进行决策[33,34]。当胎儿SMN1为1拷贝时,诊断为缺失型携带者,表型正常。当胎儿SMN1为2拷贝时,孕胎为基因型和表型正常的个体。如果父母之一基因型为[2+0]时,胎儿SMN1虽有2拷贝,胎儿仍为[2+0]型携带者个体,只有拷贝数为3时,胎儿才是正常个体。
SMN1缺失型SMA产前诊断推荐采用MLPA或qPCR技术对胎儿进行SMN1基因拷贝数分析。这两个方法均为拷贝数剂量的检测,检测范围有一定的局限性:(1)如果没有父母的基因型数据,无法检测胎儿新生的SMN1微小变异;(2)无法检测胎儿为SMN1[2+0]基因型的携带者;(3)无法检测胎儿SMN1缺失的低比例嵌合体。
需要注意的是,由于SMA以SMN1纯合缺失型为主,母源污染是导致错误产前诊断的重要根源。对绒毛组织要尽可能去除可能的母源组织,当羊水有血性污染时要进行细胞培养,无论是绒毛还是羊水均应同时以STR或SNPs为标记进行连锁分析,排除母源污染的可能。
产前诊断前的遗传咨询需告知以下要点:(1)SMA产前诊断的目的是了解孕胎携带致病基因的情况。孕胎产前诊断的可能结果包括正常个体、携带者或受累胎儿。获得产前诊断结果后胎儿的取舍由其父母知情选择。(2)SMA为常染色体隐性遗传病,父母均为携带者时,每次生育SMA患儿的再发风险为25%,男女患病机会均等。SMN1[1+0]和[2+0]基因型携带者家庭的再发风险和产前诊断策略相同。(3)产前诊断的基因检测范围仅限于检测SMN1和SMN2基因的拷贝数或某个特定的微小变异;(4)产前检测所用的方法及原理,方法的敏感度、特异度、局限性和准确度;(5)胎儿取材的种类和时间,产前诊断的价格和报告时间;(6)还要特别告知,产前诊断取材中可能会无法避免母源污染,有需要重新取材的可能;(7)获得SMA产前检测报告后需进行遗传咨询。
产前诊断后的遗传咨询主要包括:(1)针对目前检测结果是否还需进一步的检测;(2)告知产前检测结果,如为受累胎儿,告知疾病相关信息;(3)当胎儿诊断为SMA受累胎儿并选择出生时,应给予相关治疗和干预措施等医学信息和建议;(4)需要强调SMN2的拷贝数与胎儿出生后的临床表型不完全相关,SMN2拷贝数仅作为胎儿临床表型预测的参考。
胚胎植入前遗传学检测(PGT)是指将辅助生殖技术(ART)和遗传学分析技术相结合,对生育遗传病患儿高风险家庭进行胚胎活检和遗传检测,选择已知疾病不受累的胚胎植入子宫从而获得健康的子代。
(1)由于胚胎基因诊断技术仍为有创检测,PGT原则上仅针对严重致畸、致残、致死性或者治疗费用极其昂贵的遗传性疾病,大部分SMA属于此类型遗传疾病,满足单基因遗传病PGT(PGT-M)的指征;(2)基因突变携带者或先证者基因检测结果明确;(3)夫妻双方对胚胎基因诊断的流程和风险充分了解,接受并出于主观愿望进行胚胎植入前基因诊断。
(1)夫妻双方均为SMA基因突变携带者;(2)夫妻中有一方或双方血液基因组SMN1未见异常,如仅有一次SMA患儿生育或妊娠史,首先推荐产前诊断;如有2次及以上SMA患儿生育史或妊娠史,不排除生殖腺嵌合或一方为SMN1[2+0]基因型的可能性,这种情况符合PGT指征;(3)由于基因和突变类型的特殊性,携带SMN1基因突变的遗传家系进行PGT,需提供试验所需的完整核心家系成员的样本(血液、组织或者DNA等);(4)夫妻双方无不适宜实施辅助生殖术的禁忌症。
1.PGT检测的目标基因为SMA的致病基因SMN1,SMN2不在检测范围。
2.PGT技术目前大多采用碱基位点(SMN1和SMN2外显子7差异位点或微小变异位点)直接检测联合家系连锁分析的检测策略,这种策略在必需家系成员样本满足要求的条件下,理论上对SMN1突变类型并无特殊限制。缺失型突变选择SMN1与SMN2在外显子7上的差异位点c.840C/T作为检测目标,以区分SMN1纯合缺失和非纯合缺失。对于SMN1致病性微小变异的检测,由于SMN2的干扰,应用PCR技术直接检测变异位点存在一定的困难。近年来NGS在PGT上逐渐运用,大大提高了SMN1微小变异的检出率。该项技术将含有微小变异位点的PCR产物与胚胎单细胞全基因组扩增产物混合,进行全基因组测序,通过直接分析目标位点比对的碱基,可清晰地判断变异碱基所占的比例。但无论缺失突变还是微小变异,均需联合家系连锁分析,以判断胚胎基因携带突变的准确情况[35,36]。
PGT检测样本可以来源于胚胎植入前的三个阶段:(1)极体(第一极体和第二极体);(2)卵裂期单个卵裂球(发育到第3天胚胎);(3)囊胚期滋养层细胞(发育到第5至6天胚胎)。目前大多采用囊胚期进行滋养层细胞活检[37]。
如上所述,PGT-M检测包括碱基位点直接检测联合家系连锁分析。单细胞全基因组扩增产物进行位点检测的方法,可采用特异性PCR或酶切等方法。家系连锁分析主要依靠基因上下游或基因内部的特异性STR或SNP。采用NGS,可获得基因上下游充足的SNP位点,选用靠近基因上下游各三个以上有效SNP进行一代验证(尽量在基因上下游1~2Mb以内)。这种方法相对于STR分型,更利于确定染色体重组的具体位置,从而能更准确判断基因突变携带情况。目前这种检测策略被广泛用于PGT-M中[35]。
这种主要依赖家系连锁分析的检测策略可以对大部分遗传家系胚胎进行准确诊断。但对于家系不全的夫妻的胚胎检测,虽然可以借助极体或单个精子的优势作为家系连锁分析的根据[40],但由于目前所应用的单细胞扩增方法还不能完全解决等位基因脱扣(ADO)的问题,给极体和单精子突变位点的检测带来一定误差。因此对于家系不全的夫妻的胚胎检测,除PGT-M流程前的风险告知和知情同意外,移植后妊娠中期的产前诊断是必要的。
试管前遗传咨询是PGT的关键环节(咨询内容同产前诊断)。生殖科临床医生(具遗传背景)根据患者的生育力评估和家系状况,告知患者PGT流程、风险和技术的局限性,签署知情同意。
胚胎检测结束后,在胚胎移植前,夫妇依据检测报告再次进行遗传咨询,确定是否移植和移植顺序。签署移植知情同意后启动移植流程。
PGT胚胎移植后如成功妊娠,孕中期建议进行羊水细胞基因验证。新生儿发育随访与所有植入前胚胎遗传学检测策略相同。
由于SMA病情严重、人群携带率高、检测方法可靠经济、遗传咨询和产前诊断可行,2008年ACMG建议不仅有SMA家族史的夫妇应接受携带者筛查,而且所有种族和民族的一般人群都应接受SMA携带者筛查[10];2009年美国妇产科学会(ACOG)建议孕前和怀孕早期的妇女应进行SMA携带者筛查[41]。
(1)高危人群:确诊为SMA患者的家庭成员,SMA患者或携带者的配偶。(2)一般人群:目前,我国尚无SMA携带者筛查的指南,根据已发表的文献报道,中国SMA携带率为1/42~1/48[3,22,42],与美国人群携带率相差不大。近期报道中国孕妇(否认SMA家族史)SMN1杂合缺失携带率为1/56[5]。因此建议一般人群进行SMA携带者筛查。
SMA携带者筛查主要针对SMN1外显子7拷贝数,当SMN1外显子7为1个拷贝时,即为SMA携带者。SMN2不在携带者筛查范围。筛查技术包括针对性筛查SMN1的实时定量PCR和MLPA技术,而NGS常常用于包括SMA在内的多种单基因遗传病的扩展性携带者筛查。
SMA携带者筛查的时机应选择在怀孕前或怀孕的早期,以便有充裕的时间进行生育选择。检测流程一般先检测女方,如果女方为携带者,再检测男方,或男女方同时检测。双方SMN1外显子7均为1个拷贝,即均为SMA携带者,生育SMA患儿的风险为25%。
携带者筛查的遗传咨询非常重要,检测前需签署知情同意书,在受检者自愿的前提下进行:(1)筛查前的咨询中应告知SMA携带者筛查的目的、筛查范围、筛查技术的敏感性、特异性和局限性。(2)特别强调SMA携带者筛查的残余风险,即无法准确获得是否为携带者的风险:①应用任何筛查技术,均难以检测约4%的[2+0]基因型SMA携带者;②应用拷贝数筛查技术,约5%的携带SMN1微小变异的携带者[1+1d]存在残余风险;③应用NGS筛查检测出的SMN1新发变异,如不能证实其致病性,该个体尚无法确认为SMA携带者;④大约2%的SMA患者因新发变异所致,其父母的携带者筛查可能一方为阴性结果;⑤仅为生殖腺嵌合的携带者,采用血液细胞筛查可能为阴性结果。该类携带者虽然筛查显示正常,而后代仍有罹患SMA的风险。
筛查后的咨询针对携带者筛查阳性结果者,要告知生育后代的发病风险;阴性结果者需要再次告知残余风险:(1)未孕携带者应告知生育选择,主要包括产前诊断和利用植入前遗传学检测的辅助生殖;(2)已孕夫妻应建议通过胎儿的产前诊断进行生育选择;(3)携带者筛查可能发现受检者为无症状或症状轻微的晚发病的成人SMA患者,建议这类患者通过神经专业进行确诊,遗传咨询和临床管理与SMA患者相同。
SMN1是SMA基因诊断、产前诊断、植入前遗传学检测和携带者筛查的目标基因,建议拷贝数分析包括SMN1和SMN2的外显子7和8,微小变异应确认是否发生在SMN1,新发现的变异应进行致病性评估。产前诊断和植入前遗传学检测应依据先证者突变基因型选择胎儿检测的策略。基于筛查SMN1外显子7拷贝数的携带者筛查,应充分告知残余风险。SMN2基因纯合缺失不导致5q-SMA,但对确诊患者进行SMN2拷贝数分析有利于临床分型、管理和治疗等临床指导和咨询。
SMA的三级预防将成为未来的发展方向。孕妇携带者筛查配合产前诊断有效阻断了SMA患儿的出生[3,4]。SMA治疗药物的上市推动了SMA新生儿筛查,临床试验证实在出现症状前开始治疗的患者在存活率和运动里程碑达成等方面都得到了更大获益。随着NGS和生物信息学分析能力的提升,将有望通过其高通量特点,满足SMA诊断、筛查和鉴别诊断的多层面需求。
执笔者:宋昉(首都儿科研究所遗传研究室);瞿宇晋(首都儿科研究所遗传研究室);戴毅(中国医学科学院北京协和医院神经科);陈晓丽(首都儿科研究所遗传研究室);马祎楠(北京大学第一医院实验中心);朱小辉(国家妇产疾病临床医学研究中心北京大学第三医院妇产科生殖中心);王剑(上海交通大学医学院附属上海儿童医学中心医学遗传科);彭晓音(首都儿科研究所附属儿童医院神经内科);李西华(复旦大学附属儿科医院神经内科);吕俊兰(国家儿童医学中心首都医科大学附属北京儿童医院神经内科)
专家组成员(按姓氏汉语拼音排序):白晋丽(首都儿科研究所遗传研究室);曹延延(首都儿科研究所遗传研究室);陈晓丽(首都儿科研究所遗传研究室);戴毅(中国医学科学院北京协和医院神经科);杜娟(中信湘雅生殖与遗传专科医院遗传中心);高媛(山东大学生殖医学研究中心分子遗传室);何蓉(中国医科大学盛京医院临床遗传科);洪思琦(重庆医科大学附属儿童医院神经内科);黄尚志(北京协和医学院医学遗传学系);蒋宇林(中国医学科学院北京协和医院产科中心);李西华(复旦大学附属儿科医院神经内科);刘俊涛(中国医学科学院 北京协和医院产科中心);吕俊兰(国家儿童医学中心 首都医科大学附属北京儿童医院神经内科);马祎楠(北京大学第一医院实验中心);毛姗姗(浙江大学医学院附属儿童医院神经内科);潘虹(北京大学第一医院实验中心);彭晓音(首都儿科研究所附属儿童医院神经内科);强荣(西北妇女儿童医院医学遗传中心);瞿宇晋(首都儿科研究所遗传研究室);沈亦平(广西壮族自治区妇幼保健院遗传代谢中心实验室,上海儿童医学中心及波士顿儿童医院);宋昉(首都儿科研究所遗传研究室);王剑(上海交通大学医学院附属上海儿童医学中心医学遗传科);魏翠洁(北京大学第一医院儿科);熊晖(北京大学第一医院儿科);许争峰(南京市妇幼保健院遗传医学中心);闫丽盈(国家妇产疾病临床医学研究中心 北京大学第三医院妇产科生殖中心);朱小辉(国家妇产疾病临床医学研究中心 北京大学第三医院妇产科生殖中心)
所有作者均声明不存在利益冲突

























