
戈谢病(GD)是一种罕见的影响细胞糖脂类再循环的遗传性代谢病。临床主要表现为不明原因的肝脾肿大、骨痛及神经系统症状等。本例报道了一例幼年出现不明原因脾大的成年患者,曾经历脾切除仍未能明确病因。再次病情加重后出现明显活动后喘憋,查体肝大明显,最终通过酶活性检测及病理检测诊断为GD、继发肝肺综合征。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者男,25岁,因"发现脾大17年,喘憋、皮肤黄染3月,加重1个月"于2018年6月3日入住北京协和医院全科医学科(普通内科)。患者8岁起家人发现磕碰后易出现鼻衄及皮肤瘀斑,外院查体发现脾大。外院查血常规:白细胞及血小板计数轻度降低,肝功能未见异常。行骨髓穿刺活检:粒系增生欠活跃,巨核细胞以颗粒巨核细胞为主,余未见明显异常。诊断考虑"原发性巨脾",予行脾切除术,术后病理提示不能除外肉芽肿性病变。监测白细胞及血小板计数恢复正常,未进一步诊治。2018年2月患者自觉活动耐量明显下降,步行300 m即出现喘憋,夜间需高枕卧位。同时皮肤黏膜及巩膜出现黄染,伴皮肤瘙痒,否认骨痛。查血常规:白细胞23.37×109/L,血红蛋白78 g/L,血小板71×109/L;尿常规:尿蛋白(-);肝功能:白蛋白23.65 g/L,丙氨酸转氨酶(ALT)134 U/L,总胆红素67.75 μmol/L,直接胆红素39.04 μmol/L,γ谷氨酰转肽酶128 U/L;肾功能:未见异常。血氨44.9 μmol/L。乙型肝炎五项、抗丙型肝炎病毒抗体、巨细胞病毒IgM抗体及嗜异性凝集试验均(-);血CA19-9、甲胎蛋白均(-)。胸腹部CT:双肺透亮度减低,余未见明显异常;肝肿大明显(图1)。超声心动图检查:射血分数66%,轻度三尖瓣反流伴中度肺动脉高压,轻度肺动脉瓣反流,全心增大,心包少量积液。外院予以保肝、利尿治疗后呼吸困难症状略有改善。遂于2018年6月3日为进一步诊治收住本院。患者病程中否认口腔及外阴溃疡、皮疹、关节肿痛、雷诺现象等症状。既往体格发育较同龄人迟缓,但学习成绩中上等,无明显活动耐量减退,上大学期间自述可与同龄人进行同样强度长跑活动,但不喜好剧烈运动。起病以来,睡眠欠佳,大小便正常。幼时脾切除手术期间曾输血支持,余既往史和家族史均无特殊。
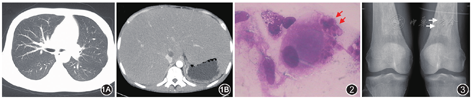
入院查体:体温36.5 ℃,心率100次/min,血压120/80 mmHg(1 mmHg=0.133 kPa),指氧饱和度(鼻导管吸氧6 L/min)86%。神智清楚,对答切题,体型偏瘦小,全身皮肤黏膜、巩膜黄染,口唇及指甲可及紫绀,无杵状指。颈部可及散在淋巴结肿大,质偏韧,活动度可。双肺呼吸音清,未闻及干湿啰音及胸膜摩擦音,心律齐,主动脉瓣听诊区可及Ⅱ度收缩期杂音,其余各瓣膜听诊区未闻及杂音。腹膨隆,肝右锁骨中线处脐下3指可及,正中线处脐下1指可及,双下肢可凹性水肿,双侧腱反射对称存在,病理征(-)。
入院后完善检查:尿常规+沉渣:尿蛋白微量,红细胞25个/μl;24 h尿蛋白:0.4 g/24 h。肝功能:白蛋白27 g/L,ALT 17 U/L,总胆红素97.2 μmol/L,直接胆红素74.8 μmol/L,γ谷氨酰转肽酶105 U/L。红细胞沉降率86 mm/1 h;超敏C反应蛋白、抗核抗体谱、自身免疫性肝炎抗体谱、抗心磷脂抗体、抗中性粒细胞胞质抗体、补体均(-);免疫球蛋白G:27.9 g/L,免疫球蛋白A:9.74 g/L;人免疫缺陷病毒抗体、快速血清反应素环状卡片试验(-);巨细胞病毒-DNA<500拷贝/ml,Epstein-Barr病毒-DNA<500拷贝/ml;降钙素原、血结核感染特异性T细胞检测、嗜异性凝集试验、布氏杆菌凝集试验、G试验均(-)。血气分析(未吸氧):pH 7.47,二氧化碳分压21 mmHg,氧分压62 mmHg,血浆碳酸氢根浓度15.3 mmol/L;肺功能:限制性通气功能障碍伴弥散功能减低。计算机断层摄影肺血管造影(CTPA):未见肺动脉栓塞表现。超声心动图检查:射血分数58%,轻度三尖瓣反流伴中度肺动脉高压,轻度肺动脉瓣反流,全心增大,心包少量积液。心脏对比增强超声:可见右心房出现气泡后第4个心动周期左房出现气泡,考虑存在肺内分流。骨髓穿刺涂片:粒系以中性分叶核粒细胞比例增高,达34%,余各阶段比例及形态大致正常。红系晚幼红细胞为主,余各阶段比例减低。红细胞大小不等,部分中心淡染区扩大。淋巴细胞及单核细胞比例形态正常。全片未见巨核细胞,血小板少见。可见戈谢细胞(图2)。骨髓活检:骨髓中纤维组织明显增生,仅见极少许造血细胞,未见巨核细胞。网织红细胞(+)。患者入院后体温正常,夜间仍无法平卧,干咳明显,余无其他不适主诉。自然状态下指氧饱和度波动于71%~80%,吸氧6 L/min,可达86%~93%。经多学科会诊考虑患者幼年起病,以不明原因脾大为突出首发表现,骨髓穿刺涂片发现戈谢细胞,考虑戈谢病可能性大。应进一步进行酶活性检测明确诊断。患者目前突出症状为呼吸困难,血气评估为Ⅰ型呼吸衰竭,CTPA未见肺栓塞,心脏对比增强超声明确存在肺内分流,结合肝脏情况,诊断考虑继发肝肺综合征。后完善β-葡糖苷酶1.9 nmol·h-1·mg·Pr-1(6.5~20.6 nmol·h-1·mgPr-1)。骨扫描:双侧第8后肋异常所见,考虑为良性病变可能性大,余骨骼未见明显异常。膝关节相:双侧股骨下端内侧骨质密度不均匀增高(图3)。建议患者开始伊米苷酶替代治疗,患者及家属讨论后因经济等方面原因拒绝替代治疗方案,返家后继续氧疗支持。2019年10月电话随诊,患者出院半年后因继发肺部感染死亡。最终诊断:戈谢病、肝肺综合征、Ⅰ型呼吸衰竭、肺动脉高压、骨髓纤维化。
戈谢病是一种常染色体隐性遗传病,常引起肝脾肿大、贫血和(或)血小板减少,以及骨痛表现,因疾病相对罕见,临床表现相对缺乏特异性,因此临床易被漏诊和误诊。
脾肿大是戈谢病常见的起病体征,如幼年出现不明原因脾大,甚至脾梗死、脾破裂等表现,应警惕遗传代谢疾病可能。
肝肺综合征是Ⅰ型戈谢病的罕见并发症,较戈谢细胞浸润引起的间质性肺疾病更为罕见,一般见于经历脾切除的戈谢病患者。
骨髓穿刺活检发现戈谢细胞可能是对疾病诊断的一个重要线索,这一诊断方法的敏感性和特异性均不高。
葡萄糖脑苷脂酶活性检测是戈谢病诊断的金标准。
替代缺陷酶治疗是指南和共识所推荐的针对Ⅰ型和Ⅲ型戈谢病的特异性治疗方案,治疗越早,效果越好,因此早期诊断和治疗对于预后非常关键。
本例患者为青年男性,隐匿起病,慢性病程,病初以无自觉症状的脾大、脾亢为主要表现,经骨髓穿刺等排查未发现血液系统恶性肿瘤的线索。经历脾切除术后,虽然脾亢表现缓解,但病情仍进一步进展,逐渐出现多系统受累的特点。甚至出现罕见的以呼吸困难为突出表现的肝肺综合征,进一步造成诊断困难。在病情较重、不具备条件进行比较大的有创检查的情况下,骨髓穿刺中发现戈谢细胞是重要的诊断突破口之一。在戈谢病患者中,通过骨髓穿刺活检发现戈谢细胞的阳性率并不高,约为30%,这也是造成病程初期误诊和漏诊率较高的重要原因之一[1]。同时这一检查的特异性并不算很高,骨髓中的单核巨噬细胞等吞噬细胞碎片或脂质代谢产物,就可能形成与戈谢细胞相似的类戈谢细胞。因此即使骨髓中发现可疑"戈谢细胞"也需要对白血病、淋巴瘤、地中海贫血、多发性骨髓瘤等多种血液病进行鉴别,并对酶活性进行检测。
戈谢病是一种常染色体隐性遗传病,是由溶酶体酶——葡糖脑苷脂酶(GBA)缺陷所致[2]。GBA是一种糖蛋白酶,主要底物是葡萄糖脑苷脂。在该病患者中,GBA缺乏导致葡萄糖脑苷脂和其他糖脂类在巨噬细胞溶酶体内蓄积。而戈谢病的临床表现即由富含脂质的巨噬细胞在脾、肝、骨髓、骨和其他组织器官中蓄积所导致。
戈谢病临床分为3个亚型,Ⅰ型最为常见,可较多见于成人,主要特点是不累及中枢神经系统,预后相对较好。Ⅱ型为爆发型,神经系统受累比较明显,预后差,患者多于2岁以内死亡。Ⅲ型可有运动失调,肌痉挛及抽搐发作表现,预后不佳,多于儿童或青春期死亡。本例患者隐匿起病,病程进展缓慢,直至成年仍未发现明显神经系统受累或者智力、认知障碍表现,因此考虑Ⅰ型戈谢病基本可明确。
在戈谢病患者中,骨骼受累是主要的临床特征之一,但骨髓纤维化罕有报道。影像学表现包括弥漫性骨质疏松、病理性骨折及骨质破坏,其中以股骨与骨盆最为常见,长骨改变以双侧股骨中下段对称性增宽,似啤酒瓶样改变最为特征。其机制尚未明确,目前怀疑戈谢细胞浸润。从机制上,戈谢病患者受累骨骼内可出现骨密度减小、骨髓浸润和骨梗死等病理改变。另外,在戈谢病体外模型中,原始造血和间充质祖细胞的增殖受损,提示有固有缺陷的参与[3,4]。如图3所示,确实发现有双侧股骨下端内侧骨质密度的不均匀增高,并且骨髓活检提示骨髓纤维化,但这些并非戈谢病典型的骨和骨髓的常见受累表现,未来尚需更多病例的总结和观察。
肝肺综合征和肺动脉高压是Ⅰ型戈谢病罕见的并发症。国际上目前仅有极少量的病例系列分析[5]。研究发现出现这些并发症的患者多于幼年诊断。幼年时这些患者均经历了脾切除术,而这一点似乎是成年后出现肝肺综合征及肺动脉高压的重要原因。通过替代治疗,无论肝肺综合征还是肺动脉高压,症状均可能得到明显缓解。目前应用替代缺陷酶治疗已经是指南和共识所推荐的对Ⅰ型和Ⅲ型戈谢病的特异性方案,治疗越早,效果越好,因此本病的早期诊断和治疗对于预后非常关键[6]。但因经济原因,本例患者未能接受替代治疗,最终因肺部感染死亡。
所有作者均声明不存在利益冲突

























