
肺动脉高压(pulmonary hypertension, PH)是指由多种异源性疾病(病因)和不同发病机制所致肺血管结构或功能改变,引起肺血管阻力和肺动脉压力升高的临床和病理生理综合征,继而发展成右心衰竭甚至死亡。近年来PH领域诊断及治疗策略不断更新,国内外在不同领域发表了PH相关指南和专家共识。为更好指导我国医师的临床实践,中华医学会呼吸病学分会肺栓塞与肺血管病学组、中国医师协会呼吸医师分会肺栓塞与肺血管病工作委员会基于当前的循证医学证据,制订了《中国肺动脉高压诊断与治疗指南(2021版)》。本指南系统评价了国内外近年来发表的PH领域系列指南和相关循证医学研究证据,增加了基于国人循证医学研究的数据,旨在进一步规范我国PH的诊断与治疗。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
肺动脉高压(pulmonary hypertension, PH)是指由多种异源性疾病(病因)和不同发病机制所致肺血管结构或功能改变,引起肺血管阻力和肺动脉压力升高的临床和病理生理综合征,继而发展成右心衰竭甚至死亡。近年来PH领域取得了许多进展,诊断及治疗策略不断更新。国内外也在不同领域发表了PH相关指南和专家共识[1, 2, 3]。
为更好指导我国医师的临床实践,中华医学会呼吸病学分会肺栓塞与肺血管病学组、中国医师协会呼吸医师分会肺栓塞与肺血管病工作委员会基于当前的循证医学证据,组织国内呼吸与危重症医学、心血管病学、风湿病学、影像学、基础医学、循证医学等领域的多学科专家,制订了《中国肺动脉高压诊断与治疗指南(2021版)》。本指南系统评价了国内外近年来发表的PH领域的系列指南和相关循证医学研究证据,增加了基于国人循证医学研究的数据,旨在进一步规范我国PH的诊断与治疗。
证据和推荐意见的评价方法采用推荐分级的评估、制定与评价(Grading of Recommendations Assessment,Development and Evaluation,GRADE)分级系统(表1、2)[4],由兰州大学循证医学中心/GRADE中国中心提供方法学支持。
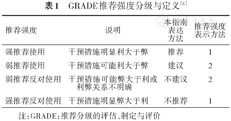
GRADE推荐强度分级与定义[4]
GRADE推荐强度分级与定义[4]
| 推荐强度 | 说明 | 本指南 表达 方法 | 推荐强度 表示方法 |
|---|---|---|---|
| 强推荐使用 | 干预措施明显利大于弊 | 推荐 | 1 |
| 弱推荐使用 | 干预措施可能利大于弊 | 建议 | 2 |
| 弱推荐反对使用 | 干预措施可能弊大于利或利弊关系不明确 | 不建议 | 2 |
| 强推荐反对使用 | 干预措施明显弊大于利 | 不推荐 | 1 |
注:GRADE:推荐分级的评估、制定与评价

GRADE证据质量分级与定义[4]
GRADE证据质量分级与定义[4]
| 质量等级 | 定义 |
|---|---|
| 高(A) | 非常确信真实值接近观察值 |
| 中(B) | 对观察值有中等程度信心:真实值有可能接近观察值,但仍存在两者不同的可能性 |
| 低(C) | 对观察值的确信程度有限:真实值可能与观察值不同 |
| 极低(D) | 对观察值几乎没有信心:真实值很可能与观察值不同 |
注:GRADE:推荐分级的评估、制定与评价
指南设计与制订步骤依据2015年《世界卫生组织指南制订手册》[5],以及2016年中华医学会发布的《制订/修订<临床诊疗指南>的基本方法及程序》[6]。
本指南小组应用临床指南研究与评估系统Ⅱ(The Appraisal of Guidelines for Research and Evaluation Ⅱ,AGREE Ⅱ)[7]对相关性比较高的指南进行评价。通过筛选,最终纳入指南35部,其中英文指南28部,中文指南7部。4名研究人员应用AGREE Ⅱ对纳入指南独立进行评价,预实验结果显示4名研究人员对评分条目的理解一致性较好。经过评价每部指南各领域的得分情况,对每部指南是否推荐使用的结果显示,13部(37.1%)指南被推荐使用,15部(42.9%)被推荐修订后使用,7部(20%)不推荐使用(表3)。
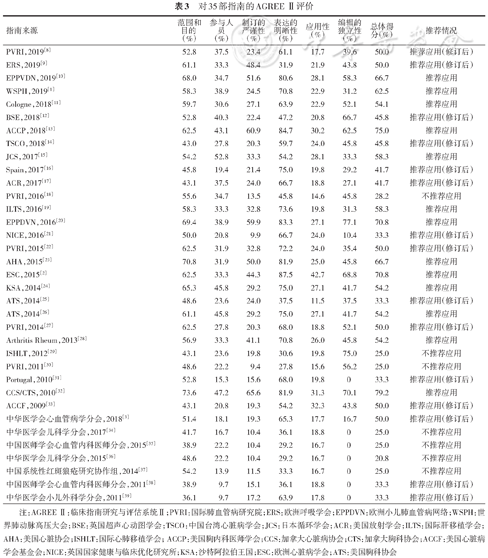
对35部指南的AGREE Ⅱ评价
对35部指南的AGREE Ⅱ评价
| 指南来源 | 范围和目的 (%) | 参与人员 (%) | 制订的严谨性(%) | 表达的明晰性(%) | 应用性 (%) | 编辑的独立性(%) | 总体得分(%) | 推荐情况 |
|---|---|---|---|---|---|---|---|---|
| PVRI,2019[8] | 52.8 | 37.5 | 23.4 | 61.1 | 17.7 | 39.6 | 50.0 | 推荐应用(修订后) |
| ERS,2019[9] | 61.1 | 33.3 | 48.4 | 31.9 | 21.9 | 43.8 | 50.0 | 推荐应用(修订后) |
| EPPVDN,2019[10] | 68.0 | 34.7 | 51.6 | 80.6 | 28.1 | 58.3 | 66.7 | 推荐应用 |
| WSPH,2019[1] | 58.3 | 38.9 | 24.5 | 70.8 | 22.9 | 31.2 | 62.5 | 推荐应用 |
| Cologne,2018[11] | 59.7 | 30.6 | 27.1 | 63.9 | 22.9 | 52.1 | 54.1 | 推荐应用 |
| BSE,2018[12] | 52.8 | 40.3 | 22.4 | 47.2 | 20.8 | 66.7 | 45.8 | 推荐应用(修订后) |
| ACCP,2018[13] | 62.5 | 43.1 | 60.9 | 84.7 | 30.2 | 62.5 | 75.0 | 推荐应用 |
| TSCO,2018[14] | 43.0 | 27.8 | 20.3 | 59.7 | 24.0 | 45.8 | 45.8 | 推荐应用(修订后) |
| JCS,2017[15] | 54.2 | 52.8 | 33.3 | 54.2 | 28.1 | 33.3 | 58.3 | 推荐应用 |
| Spain,2017[16] | 45.8 | 19.4 | 21.4 | 75.0 | 19.8 | 29.2 | 41.7 | 推荐应用(修订后) |
| ACR,2017[17] | 43.1 | 37.5 | 24.0 | 66.7 | 18.8 | 27.1 | 41.7 | 推荐应用(修订后) |
| PVRI,2016[18] | 55.6 | 34.7 | 13.5 | 45.8 | 14.6 | 45.8 | 28.2 | 不推荐应用 |
| ILTS,2016[19] | 58.3 | 33.3 | 32.8 | 73.6 | 19.8 | 31.3 | 58.3 | 推荐应用 |
| EPPDVN,2016[20] | 69.4 | 38.9 | 59.9 | 83.3 | 27.1 | 77.1 | 70.8 | 推荐应用 |
| NICE,2016[21] | 50.0 | 20.8 | 9.9 | 66.7 | 24.0 | 10.4 | 33.3 | 推荐应用(修订后) |
| PVRI,2015[22] | 62.5 | 31.9 | 32.8 | 72.2 | 24.0 | 35.4 | 50.0 | 推荐应用(修订后) |
| AHA,2015[23] | 70.8 | 31.9 | 50.0 | 81.9 | 25.0 | 45.8 | 66.7 | 推荐应用 |
| ESC,2015[2] | 62.5 | 33.3 | 44.3 | 87.5 | 42.7 | 68.8 | 70.8 | 推荐应用 |
| KSA,2014[24] | 65.3 | 45.8 | 29.2 | 75.0 | 27.1 | 41.7 | 54.2 | 推荐应用 |
| ATS,2014[25] | 48.6 | 23.6 | 24.0 | 37.5 | 11.5 | 37.5 | 33.3 | 推荐应用(修订后) |
| ATS,2014[26] | 61.1 | 45.8 | 29.2 | 75.0 | 27.1 | 41.7 | 54.2 | 推荐应用 |
| PVRI,2014[27] | 62.5 | 27.8 | 20.3 | 68.0 | 18.8 | 52.1 | 50.0 | 推荐应用(修订后) |
| Arthritis Rheum,2013[28] | 56.9 | 33.3 | 41.1 | 70.8 | 26.0 | 45.8 | 54.2 | 推荐应用 |
| ISHLT,2012[29] | 43.1 | 23.6 | 19.8 | 30.6 | 19.8 | 75.0 | 25.0 | 不推荐应用 |
| PVRI,2011[30] | 48.6 | 22.2 | 9.4 | 27.8 | 15.6 | 56.2 | 25.0 | 不推荐应用 |
| Portugal,2010[31] | 52.8 | 15.3 | 15.6 | 68.0 | 19.8 | 0 | 33.3 | 推荐应用(修订后) |
| CCS/CTS,2010[32] | 73.6 | 47.2 | 65.6 | 81.9 | 31.3 | 70.1 | 79.2 | 推荐应用 |
| ACCF,2009[33] | 43.1 | 20.8 | 19.3 | 54.2 | 32.3 | 43.8 | 50.0 | 推荐应用(修订后) |
| 中华医学会心血管病学分会,2018[3] | 51.4 | 18.1 | 19.3 | 65.3 | 17.7 | 16.7 | 50.0 | 推荐应用(修订后) |
| 中华医学会儿科学分会,2017[34] | 41.7 | 16.7 | 10.4 | 36.1 | 18.8 | 0 | 25.0 | 不推荐应用 |
| 中国医师学会心血管内科医师分会,2015[35] | 38.9 | 22.2 | 10.4 | 29.2 | 16.7 | 0 | 25.0 | 不推荐应用 |
| 中华医学会儿科学分会,2015[36] | 48.6 | 22.2 | 10.4 | 29.2 | 16.7 | 0 | 20.8 | 不推荐应用 |
| 中国系统性红斑狼疮研究协作组,2014[37] | 54.2 | 13.9 | 11.5 | 33.3 | 16.7 | 0 | 25.0 | 不推荐应用 |
| 中国医师学会心血管内科医师分会,2011[38] | 38.9 | 9.7 | 15.1 | 36.1 | 18.8 | 0 | 33.3 | 推荐应用(修订后) |
| 中华医学会小儿外科学分会,2011[39] | 36.1 | 9.7 | 17.2 | 63.9 | 17.8 | 0 | 33.3 | 推荐应用(修订后) |
注:AGREE Ⅱ:临床指南研究与评估系统Ⅱ;PVRI:国际肺血管病研究院;ERS:欧洲呼吸学会;EPPDVN:欧洲小儿肺血管病网络;WSPH:世界肺动脉高压大会;BSE:英国超声心动图学会;TSCO:中国台湾心脏病学会;JCS:日本循环学会;ACR:美国放射学会;ILTS:国际肝移植学会;AHA:美国心脏协会;ISHLT:国际心肺移植学会; ACCP:美国胸内科医师学会;CCS:加拿大心脏病协会;CTS:加拿大胸科协会;ACCF:美国心脏病学会基金会;NICE:英国国家健康与临床优化研究所;KSA:沙特阿拉伯王国;ESC:欧洲心脏病学会;ATS:美国胸科协会
本指南的报告和撰写参考卫生保健实践指南的报告条目(reporting items for practice guidelines in healthcare, RIGHT)[40]。
本指南的整体技术路线见图1。
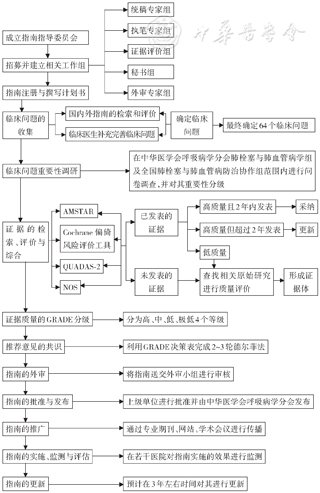
注:AMSTAR:系统评价、荟萃分析方法学质量评价工具; QUADAS:诊断准确性研究的质量评价工具;NOS:纽卡斯尔‑渥太华文献质量评价量表;GRADE:推荐分级的评估、制定与评价
1.指南注册与计划书的撰写:本指南已在国际实践指南注册平台(http://www.guidelines-registry.cn)进行注册(注册号:IPGRP-2019CN050),读者可联系指南发起组织索要指南的计划书。
2.指南使用者与目标人群:本指南供中国内科(呼吸与危重症医学科、心血管科、风湿免疫科等)、外科(胸外科、心外科等)、儿科、急诊科、药剂科、影像科及其他与PH诊疗和管理相关学科的专业人员使用。指南推荐意见的应用目标人群为中国PH患者。
3.指南工作组:本指南成立了多学科专家工作组,包括呼吸与危重症医学科、心血管科、风湿免疫科、儿科、胸外科、心外科、影像科、基础医学、循证医学等。工作组具体分为5个:统稿专家组、执笔专家组、证据评价组、秘书组、外审组。
4.利益冲突声明:本指南工作组成员均填写了利益冲突声明表,不存在与本指南撰写内容直接相关的利益冲突。
5.临床问题遴选和确定:通过系统检索PH领域已经发表的指南和系统评价,第1轮收集了408个临床问题,对其进行去重合并后,邀请临床医师对其进一步修改和补充,第2轮形成139个临床问题。临床问题按其重要性分为1~7分。在中华医学会呼吸病学分会肺栓塞与肺血管病学组、中国医师协会呼吸医师分会肺栓塞与肺血管病工作委员会(46位专家)和全国肺栓塞与肺血管病防治协作组(556家医院的2 954名医师)范围内进行问卷调查并对其进行重要性分级。基于调查结果,纳入最终本指南需解决的64个临床问题。
6.证据检索:针对最终纳入的临床问题与结局指标,按照“人群、干预、对照和结局”对其进行解构,并根据解构的问题:(1)检索Medline、Embase、Cochrane Library、Epistemonikos、CBM、万方和CNKI等数据库,纳入系统评价、Meta分析、网状Meta分析,检索时间为建库至2020年6月30日;(2)检索Uptodate、DynaMed、CBM、万方和CNKI等数据库,纳入原始研究(包括随机对照试验、队列研究、病例对照研究、病例系列、流行病学调查等),检索时间为建库至2020年6月30日;(3)检索策略:根据前期筛选的临床问题确定相应的检索策略。
7.证据评价:证据评价与分级小组使用评估系统评价的测量工具(assessment of multiple systematic reviews,AMSTAR)[41]对纳入的系统评价、Meta分析、网状Meta分析进行方法学质量评价,使用Cochrane偏倚风险评价工具(针对随机对照试验)[42]、诊断准确性研究的质量评价工具(quality assessment of diagnostic accuracy studies-2,QUADAS-2)[43]、纽卡斯尔-渥太华文献质量评价量表(Newcastle-Ottawa scale,NOS)[44]对相应类型的原始研究进行方法学质量评价;使用GRADE方法对证据体进行评价和推荐意见进行分级。
8.推荐意见形成:专家小组基于证据评价与分级小组提供的PH诊断、治疗安全性和有效性的国内外循证医学证据,经过3轮德尔菲法和2轮面对面专家小组会,对推荐意见达成共识,最终形成54项推荐意见。
本指南推荐内容的主要特点:(1)首次将欧美指南的格式和表述方法与中国临床实际情况结合起来。(2)提出符合中国医师临床实践的PH诊断及治疗流程。(3)重视PH患者的随访与管理。根据患者治疗前的基础状态和短期治疗后的临床指标来判断病情和评估预后,以利于及时调整治疗方案。(4)提出建立专业的PH中心、完善PH患者转诊制度。
本指南表述方式的特点:(1)指南中将每条推荐意见单独列出,以便读者迅速发现与阅读;(2)推荐意见后清楚标记GRADE分级符号,明确区分推荐意见的方向与强度,并应用“建议”或“推荐”字样,以便读者进一步明确推荐级别;(3)清晰显示主题、标题、推荐意见、推荐意见说明、相关证据汇总等格式,备注相应的说明文字,以解释该条推荐意见的理由;证据总结的目的是清楚呈现支持该条推荐意见的主要证据与来源。
9.指南更新:指南工作组计划在3年左右时间对本指南进行更新。更新方法按照国际指南更新流程进行[45, 46]。
10.传播与实施:指南发布后,指南工作组将主要通过以下方式对指南进行传播与推广:(1)在相关学术期刊发表;同期编写指南精简版及详细解读手册;(2)出版指南相对应的医生版、护士版和基层版,在相关学术会议中进行解读;(3)在国内不同区域、不同学科组织指南推广专场,确保临床医师充分了解并正确应用本指南;(4)通过微信、网络和其他媒体进行推广。
PH是指海平面、静息状态下,经右心导管检查(right heart catheterization,RHC)测定的肺动脉平均压(mean pulmonary artery pressure, mPAP)≥25 mmHg(1 mmHg=0.133 kPa)[2]。正常成年人静息状态下mPAP为(14.0±3.3)mmHg,其上限不超过20 mmHg[47]。mPAP在21~24 mmHg曾被定义为临界性PH [48],在2018年第六届世界肺动脉高压大会(World Symposium on Pulmonary Hypertension,WSPH)上,有专家建议将PH血流动力学诊断标准修改为mPAP>20 mmHg[49],但由于存在广泛争议,目前我国也尚缺乏针对mPAP在21~24 mmHg患者的相关研究,因此本指南没有采纳这一诊断标准。但针对mPAP在21~24 mmHg的人群,特别是存在结缔组织病(connective tissue disease,CTD)、血栓栓塞性疾病、特发性肺动脉高压(idiopathic pulmonary arterial hypertension,IPAH)家族史等情况的人群,确实有必要重视其筛查、随访与管理,建立我国此类人群数据库,开展多中心临床研究[50]。
PH的血流动力学定义及分类见表4。

肺动脉高压(PH)的血流动力学分类
肺动脉高压(PH)的血流动力学分类
| 血流动力学分类 | 分类标准 | 临床分类 |
|---|---|---|
| 毛细血管前肺动脉高压 | mPAP≥25 mmHg且 PAWP≤15 mmHg | 动脉性肺动脉高压;肺部疾病和/或低氧所致肺动脉高压;慢性血栓栓塞性肺动脉高压和/或其他肺动脉阻塞性肺动脉高压;未明和/或多因素所致肺动脉高压 |
| 毛细血管后肺动脉高压 | 左心疾病所致肺动脉高压;未明和/或多因素所致肺动脉高压 | |
| 单纯性 | mPAP≥25 mmHg且PAWP>15 mmHg且PVR≤3 WU | |
| 混合性 | mPAP≥ 25 mmHg且PAWP>15 mmHg且PVR>3 WU |
注:mPAP:肺动脉平均压;PAWP:肺动脉楔压;PVR:肺血管阻力。1 mmHg =0.133 kPa
临床上将PH分为5大类(表5):(1)动脉性PH(pulmonary arterial hypertension,PAH);(2)左心疾病所致PH;(3)肺部疾病和(或)低氧所致PH;(4)慢性血栓栓塞性PH(chronic thromboembolic pulmonary hypertension,CTEPH)和(或)其他肺动脉阻塞性病变所致PH;(5)未明和(或)多因素所致PH。

肺动脉高压(PH)的临床分类
肺动脉高压(PH)的临床分类
| 分类 | 亚类 |
|---|---|
| 1.动脉性肺动脉高压(PAH) | 1.1特发性肺动脉高压(IPAH) 1.2遗传性肺动脉高压(HPAH) 1.3药物和毒物相关肺动脉高压 1.4疾病相关的肺动脉高压 1.4.1 结缔组织病 1.4.2 HIV感染 1.4.3 门脉高压 1.4.4 先天性心脏病 1.4.5 血吸虫病 1.5 对钙通道阻滞剂长期有效的肺动脉高压 1.6 具有明显肺静脉/肺毛细血管受累(肺静脉闭塞病/肺毛细血管瘤病)的肺动脉高压 1.7 新生儿持续性肺动脉高压(PPHN) |
| 2.左心疾病所致肺动脉高压 | 2.1 射血分数保留的心力衰竭 2.2 射血分数降低的心力衰竭 2.3 瓣膜性心脏病 2.4 导致毛细血管后肺动脉高压的先天性/获得性心血管病 |
| 3.肺部疾病和/或低氧所致肺动脉高压 | 3.1 阻塞性肺疾病 3.2 限制性肺疾病 3.3 其他阻塞性和限制性并存的肺疾病 3.4 非肺部疾病导致的低氧血症 3.5 肺发育障碍性疾病 |
| 4.慢性血栓栓塞性肺动脉高压和/或其他肺动脉阻塞性病变所致肺动脉高压 | 4.1 慢性血栓栓塞性肺动脉高压(CTEPH) 4.2 其他肺动脉阻塞性疾病:肺动脉肉瘤或血管肉瘤等恶性肿瘤、肺血管炎、先天性肺动脉狭窄、寄生虫(包虫病) |
| 5.未明和/或多因素所致肺动脉高压 | 5.1 血液系统疾病(如慢性溶血性贫血、骨髓增殖性疾病) 5.2 系统性和代谢性疾病(如结节病、戈谢氏病、糖原储积症) 5.3复杂性先天性心脏病 5.4 其他(如纤维性纵隔炎) |
全球范围内有关PH流行病学相关文献报道很少。在英国, PH患病率为97/百万,其中女性/男性比例为1.8∶1。不同类型PH流行病学资料不同[2]。左心疾病、肺部疾病和/或低氧所致PH是临床工作中最常见的类型,但相关的人口统计学资料、临床病程等数据非常有限,应建立相关注册登记研究,有助于对此类疾病的了解。
多项国际注册登记研究中关于PAH的流行病学数据显示[51, 52],成人PAH 人群发病率约2.4/百万人年,患病率约15/百万。在欧洲,PAH人群发病率和患病率分别为5/百万人年~10/百万人年、15/百万~60/百万。PAH中以先天性心脏病(congenital heart diseace,CHD)相关PAH、遗传性PAH(heritable pulmonary arterial hypertension,HPAH)、药物和毒物相关PAH(drug-induced pulmonary hypertension,DPAH)常见,相关因素 PAH 中则以CTD最为常见。
IPAH目前病因不明,注册登记研究报道IPAH最低患病率约为5.9/百万。从1981年美国NIH数据来看,IPAH的平均发病年龄为36岁,以女性为主。近期的注册研究中,诊断PAH的中老年人有增多趋势,对这部分患者应注意除外左心疾病或慢性肺病所致PH的可能[52]。
一些危险因素可能对PAH疾病发生发展起到诱发或促进作用。根据与PAH发生的相关程度和致病性,将危险因素分为肯定相关及可能相关[53, 54, 55]。
在缺乏靶向药物的传统治疗时代,IPAH自然预后差,中位生存期仅2.8年, 1、3 和 5 年生存率分别为 68%、48%和34%[56]。随着靶向药物治疗进展,IPAH预后明显改善。美国REVEAL注册研究显示,自靶向药物广泛应用于临床以后(自2001年11月之后),PAH患者1、3、5和7年的Kaplan-Meier生存率分别为85%、68%、57%和49%。日本的研究显示经过靶向药治疗, IPAH患者长期生存显著提高,5年生存率达96%,10年生存率达78%。2011年我国研究表明,IPAH 的 1、3 年生存率分别为92.1%、75.1%,与发达国家报道相近[57]。
CHD-PAH是指由体-肺分流型CHD所引起的肺动脉压力升高,是我国PAH最常见的原因,诸多CHD患者因罹患PAH而失去手术机会。CHD-PAH流行病学资料较少,尚无成人CHD患者PAH发病率的报道,欧洲一项注册登记研究提示,成年CHD患者中PAH 患病率为5%~10%[58]。
PAH是CTD的常见并发症,美国REVEAL注册研究显示CTD相关PAH占所有PAH患者的25.3%。我国系统性红斑狼疮(systemic lupus erythematosus,SLE)、干燥综合征(Sjögren syndrome, SS)、混合性结缔组织病(mixed connective tissue disease,MCTD)以及系统性硬化症(systemic sclerosis,SSc)是最常引起PAH的CTD[59]。中国系统性红斑狼疮协作组注册研究结果显示SLE-PAH患病率为3.8%[60]。我国对SLE近30年的死因分析发现,SLE-PAH是导致SLE患者死亡的第3位原因。文献报道欧美SSc占CTD-PAH患者总数的67.7%~76%[51, 61, 62],而我国因拥有SLE的高发人群,故以SLE-PAH更为常见,占比约为49%[59]。
门脉高压相关性肺动脉高压(portopulmonary hypertension, PoPH)是门脉高压合并肺血流量增加或肺血管重塑而引起的PH,可伴或不伴肝脏疾病。PoPH在各种原因所致的PAH中占5%~10%[63],在门脉高压患者中发病率为1%~5%[64]。PoPH通常发生在诊断门脉高压4~7年后,门脉高压和肝硬化的严重程度与PoPH的发生无关[65], 但会影响其预后[66]。
HIV感染是PAH的危险因素之一,HIV感染患者PAH患病率在0.5%左右,显著高于普通人群PAH患病率[51, 67]。抗逆转录病毒疗法应用以来,HIV-PAH的发病率并无明显改变[51]。研究表明,HIV-PAH多发生在25~44岁年龄段,女性的发生率更高[68]。
左心疾病是PH的最常见原因,左心疾病所致PH的流行病学数据仍不清楚,不同的文献因为肺动脉压力的检测方法不同、PH的定义不同及RHC诊断标准尚未统一,所以在射血分数保留的心力衰竭(heart failure with preserved injection fraction, HFpEF)与射血分数降低的心力衰竭(heart failure with reduced injection fraction, HFrEF)中得出的患病率具有广泛差异性[69]。其中PH-HFpEF为36%~83%,PH-HFrEF为40%~75%[70]。
慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)是该类PH最常见的病因,研究发现COPD相关PH的患病率差别较大,为20%~91%不等[71, 72, 73],而肺纤维化合并肺气肿(combined pulmonary fibrosis and emphysema,CPFE)并发PH的患病率更高,可以达到30%~50%[74, 75, 76]。间质性肺疾病(interstitial lung disease,ILD)相关PH的流行病学数据大多数来自于对特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)的研究,8%~15%的IPF患者在初次检查时即发现mPAP≥25 mmHg,而在疾病晚期和终末期PH的患病率更是高达30%~50%,甚至>60%[77, 78, 79]。
PAH的病理改变主要累及远端肺小动脉,其特征性表现为:肺动脉内膜增殖伴炎症反应、内皮间质化,甚至形成向心性或偏心性改变,中膜肥厚及持续的收缩、外膜纤维化、基质重塑以及肺小血管周围炎症浸润而导致其增厚、滋养血管屈曲增生形成丛状病变;还可见病变远端扩张和原位血栓形成,从而导致肺动脉管腔进行性狭窄、闭塞[87]。近年来研究还发现肺静脉也会出现血管重塑,出现“动脉化”表现,参与PAH的发生;支气管动脉因为“血管分流”会出现管壁增厚和管腔扩大等表现。
PH发病机制复杂,是多因素、多环节共同作用的结果,包括外因(低氧、烟草、粉尘、其他理化生物因素等)、内因(遗传、发育、结构、疾病等)及交互因素(微生态、感染、免疫、药物等)。多种血管活性分子[内皮素、血管紧张素Ⅱ、前列环素、一氧化氮(nitric oxide, NO)、一氧化碳、硫化氢及二氧化硫、雌激素等]、多种离子通道(钾离子通道、钙离子通道、锌离子通道及新型阳离子通道)、多条信号通路[低氧诱导因子/TRPC通路、MAPK通路、Rho/ROCK通路、PI3K/AKT通路、BMP/TGF-β通路、核因子κB(NF-κB)通路和Notch通路]参与PH疾病的发生发展。
肺动脉压力的高低取决于肺血流量和肺血管阻力(pulmonary vascular resistance, PVR)的综合效应。PVR主要由肺小动脉、肺毛细血管和肺静脉阻力构成。任何可导致肺血流量增加和或肺血管阻力升高的结构和功能异常的因素均可引发PH。肺动脉压力升高导致右心后负荷增加,从而引起右心室肥厚、扩张、功能不全,最终出现右心衰竭[88, 89]。
左心疾病所致PH主要由于左心收缩、舒张功能障碍和/或左心瓣膜疾病引起的肺动脉压力异常升高所致, 其病理生理特征为左心充盈压升高,肺静脉回流受阻,肺静脉压力升高,从而继发肺动脉压力升高。
肺部疾病和/或低氧所致PH是一类由于肺实质或间质长期破坏、缺氧以及继发的肺血管床损害所导致的PH。其病理生理学机制涉及低氧相关肺血管收缩/重塑、血管内皮及平滑肌功能障碍、炎症、高凝状态等多个环节[90, 91]。
CTEPH致病因素较多,发病机制复杂,部分患者是急性PTE的一种远期并发症。急性PTE后血栓不完全溶解并发生机化,导致PVR持续增加,引起肺血管重塑,最终导致右心功能衰竭[92]。
基因突变与部分PAH患者发病相关,HPAH均为单基因常染色体显性遗传。目前已知9个致病基 因:BMPR2、BMP9、ALK1、Endoglin、SMAD9、BMPR1B、TBX4、CAV1 和 KCNK3,可解释50%~80% 的HPAH和 20%~50% 的散发型IPAH患者的病因[93, 94, 95]。我国学者新近发现IPAH人群PTGIS基因突变(6.1%),合并该基因突变者对伊洛前列环素治疗反应更敏感[96]。
BMPR2是PAH最常见的致病基因,可解释75%的家族HPAH及25%的IPAH散发病例。中国人群中BMPR2突变比例在HPAH和 IPAH 分别为 53% 和 15%[97]。BMPR2编码骨形成蛋白2型受体,在调控血管增殖中起到重要作用。与不携带突变的患者相比,携带BMPR2 突变的IPAH/HPAH患者发病更早,临床表型更严重,预后更差[98]。ALK1和Endoglin是遗传性出血性毛细血管扩张症(hereditary hemorrhagic telangiectasia,HHT)相关PAH最主要的致病基因[93]。在肺静脉闭塞病(pulmonary veno-occlusive disease,PVOD)/肺毛细血管瘤病(pulmonary capillary haemangiomatosis,PCH)家族中,发现常染色体隐性遗传基因突变,全基因组测序显示,在所有家族性PVOD/PCH,以及25%组织学确诊的散发PVOD/PCH病例中存在EIF2AK4突变[99]。对于临床疑似 PVOD/PCH患者,如检出EIF2AK4 双等位基因突变,有助于确诊PVOD/PCH[95]。
PH的临床症状缺乏特异性,主要表现为进行性右心功能不全的相关症状,常为劳累后诱发,表现为疲劳、呼吸困难、胸闷、胸痛和晕厥,部分患者还可表现为干咳和运动诱发的恶心、呕吐。晚期患者静息状态下可有症状发作。随着右心功能不全的加重可出现踝部、下肢甚至腹部、全身水肿。导致PH的基础疾病或伴随疾病也会有相应的临床表现[2, 100]。部分患者的临床表现与PH的并发症和肺血流的异常分布有关,包括咯血、声音嘶哑、胸痛等。严重肺动脉扩张可引起肺动脉破裂或夹层。
PH心电图可表现为肺性P波、QRS电轴右偏、右室肥厚、右束支传导阻滞、QTc间期延长等。心电图对PH诊断的敏感性低,正常心电图并不能排除PH。异常心电图多见于严重的PH。
右室肥厚有助于初诊PH患者的诊断并对预后具有预测价值,但用于PH筛查的敏感性和特异性低[101]。QRS波群和QTc间期延长提示病情严重[102]。疾病晚期可见室上性心律失常,尤其是心房扑动和心房颤动,室性心律失常少见。房性心律失常影响心输出量,加重病情。
PH患者胸部X线可见肺动脉段凸出,中心肺动脉扩张,与周围肺动脉纤细或截断形成鲜明对比,表现为“残根”征,以及右心房和右心室扩大的征象。
X线胸片有助于筛查PH的病因,如左心疾病、肺部疾病、先天性心脏病和栓塞性疾病等在X线胸片上具有相应的影像学特征。PH的严重程度与胸片异常程度并无相关,正常的X线胸片不能排除PH。
肺功能检查在PH的病因诊断中具有较高价值,对于肺部疾病所致PH,根据第1秒用力肺活量(forced expiratory volume in one second, FEV1)、用力肺活量(forced vital capacity, FVC)、肺总量(total lung capacity, TLC)、一氧化碳弥散量(carbon monoxide diffusing capacity,DLco)可以鉴别阻塞性、限制性以及混合性通气功能障碍的肺部疾病。胸廓畸形、胸膜增厚与ILD相关PH在肺功能的表现上相似,可以表现为肺容积的减少。PAH由于血管的张力增高,肺组织僵硬度增加,可表现为轻度限制性通气功能障碍,同时肺小动脉扩张压迫终末呼吸道或肺泡也可引起轻度气道阻塞。大部分PAH患者的弥散功能表现为轻或中度下降。
阻塞性气道疾病及神经肌肉疾患可能表现为低氧血症及高碳酸血症。如出现与疾病程度不相符的低氧血症需考虑到动静脉分流的情况。轻症PAH的动脉血气分析可完全正常,病情严重者可能存在过度通气,表现为二氧化碳分压下降及低氧血症。
肺功能测定和动脉血气分析不仅可以帮助发现潜在的气道或肺部疾病,还和PAH的严重程度相关。IPAH患者如DLco显著降低(<45%预测值)往往提示心输出量明显降低,预示预后不良[103, 104]。IPAH患者二氧化碳分压值越低,说明过度通气越严重,预后越差,而氧分压和预后无明确相关性[105]。
超声心动图可用于PH诊断筛查、病因鉴别和心功能评价。
根据静息状态下超声心动图测量的三尖瓣反流峰值流速(tricuspid regurgitation velocity,TRV)和其他指标可以评估PH的可能性(表6),用低、中、高度可能表示。根据临床表现和超声心动图评估的PH可能性判断是否需行RHC。除TRV外,其他提示PH的指标参见表7。对于有症状的患者,可依据超声心动图PH的可能性作进一步评估[106, 107]。

可疑肺动脉高压(PH)患者超声心动图诊断PH的可能性
可疑肺动脉高压(PH)患者超声心动图诊断PH的可能性
| 三尖瓣反流峰值流速(m/s) | 存在其他支持PH的超声心动图征象 | PH的可能性 |
|---|---|---|
| ≤2.8或测不出 | 无 | 低 |
| ≤2.8或测不出 | 有 | 中 |
| 2.9~3.4 | 无 | 中 |
| 2.9~3.4 | 有 | 高 |
| >3.4 | 不需要 | 高 |

其他支持肺动脉高压(PH)的超声心动图征象
其他支持肺动脉高压(PH)的超声心动图征象
| A:心室a | B:肺动脉a | C:下腔静脉和右心房a |
|---|---|---|
| 右心室/左心室内径比>1.0 | 多普勒右室流出道加速时间<105 ms, 和/或收缩中期切迹 | 下腔静脉直径>21 mm 伴吸气时塌陷(深吸气时塌陷率<50%或平静呼吸时塌陷率<20%) |
| 室间隔扁平(收缩期和/或舒张期左室偏心指数>1.1) | 舒张早期肺动脉反流速度>2.2 m/s | 收缩末期右心房面积>18 cm2 |
| 主肺动脉直径>25 mm |
注:a至少满足A、B、C三类指标中的两项,方可说明存在支持PH的超声心动图征象
超声心动图有助于鉴别PH的病因,如CHD、左心疾病等。经食道超声对于某些CHD的诊断更为准确。
超声心动图对于心脏功能评价具有较好的价值,如可根据三尖瓣环收缩期位移(tricuspid annular plane systolic excusion,TAPSE)、右室心肌做功指数(Tei index,Tei指数)、左心室偏心指数、右心房面积等评估患者的右心功能,并可预测预后。
核素肺通气/灌注(ventilation/perfusion,V/Q)显像是判断PH患者是否存在肺动脉狭窄或闭塞性病变(包括栓塞性疾病等)的重要检查手段。如果存在呈肺段分布的灌注缺损且与通气显像不匹配,则需要考虑肺动脉狭窄/闭塞性病变的可能性。PAH的肺V/Q显像可能正常,也可能存在非肺段性灌注缺损。
筛查CTEPH应用V/Q显像比CT肺动脉造影(computer tomography pulmonary angiography,CTPA)敏感性高[108],正常或低度可能V/Q显像可基本排除CTEPH(敏感性90%~100%、特异性94%~100%)。V/Q显像易出现假阳性,尤其存在严重心肺部疾病时,需要结合其他检查进行鉴别。
CT可显示右心室和右心房扩大、主肺动脉扩张,并可通过测量主肺动脉与升主动脉直径比来评估PH可能性。高分辨CT(high resolution CT,HRCT)还有助于PH病因筛查,肺部疾病所致PH患者HRCT可检出肺气肿、肺大疱、肺纤维化等肺部病变,PVOD/PCH患者HRCT可发现弥漫性小叶中心性磨玻璃结节、小叶间隔增厚、纵隔淋巴结肿大等征象。
CTPA是诊断肺血管病的重要检查手段,对制定CTEPH的治疗方案也非常重要,可为肺动脉血栓内膜剥脱术(pulmonary thromboendarterectomy,PEA)提供影像学依据。CTEPH常见的CTPA征象包括:肺动脉完全阻塞,肺动脉内条带影、网状充盈缺损,以及肺动脉管壁不规则增厚等[109, 110]。由于CT技术的发展,CTPA诊断肺血管病的敏感性和特异性也越来越高,可部分替代肺动脉造影检查[111, 112]。
肺动脉造影主要用于了解肺血管形态和血流灌注情况,是PTE的“参比”诊断标准,也常用于其他肺血管堵塞、狭窄、闭塞和肺动静脉畸形等肺血管病变的鉴别。CTEPH患者大多需行肺动脉造影检查, 以判断能否从PEA或球囊肺动脉成形术(balloon pulmonary angioplasty, BPA)中获益。
心血管磁共振(cardiac magnetic resonance, CMR)成像可直接评价右心室大小、形态和功能,并可无创评估血流量,包括心输出量、每搏输出量和右心室质量。MR血管造影对导致肺血管堵塞的病因鉴别可能有帮助,特别适用于孕妇或对碘造影剂过敏者[113]。
由于CMR具有无创、可重复的特点,且对右心功能的评估与RHC相比具有较高的一致性,因而可作为PAH患者基线和随访时对病情严重性判断的手段[114, 115]。
血液学检查主要用于筛查PH的病因和评价器官损害情况。
风湿免疫病相关自身抗体、肝炎标志物、HIV抗体等是特定PH类型的重要标志。血常规检查异常需要警惕各类血液系统疾病(如白血病、贫血、红细胞增多症、骨髓增生异常综合征、多发性骨髓瘤等)、结缔组织疾病以及慢性缺氧性疾病(红细胞及血红蛋白代偿性升高)等。肝功能异常(主要是转氨酶和胆红素)需要考虑门脉高压、药物损伤、血液系统疾病及心衰等原因。对于原因不明的儿童PH患者,需检测同型半胱氨酸及血、尿有机酸代谢以明确是否存在代谢性疾病(如甲基丙二酸尿症等)。
CTEPH患者需要行易栓症筛查(包括遗传性和获得性),特别是抗磷脂抗体、狼疮抗凝物、抗β2 糖蛋白1抗体。
所有PH患者在初诊及随访过程中需要测定血液脑利钠肽(BNP)或N末端脑利钠肽前体(NT-proBNP),用于评估病情及指导治疗。
腹部超声可以了解腹部脏器的结构和功能,为PH的病因筛查提供依据。腹部超声可以确诊但不能完全排除门脉高压,也可以为右心衰竭提供线索,如肝脾肿大、肝瘀血、腹水以及肝静脉、门静脉扩张等。
(十一)右心导管检查和急性血管反应试验
RHC是诊断和评价PH的标准方法,通过RHC可获得血流动力学数据,包括右房压、右室压(收缩压、舒张压和平均压)、肺动脉压力(收缩压、舒张压和平均压)、肺动脉楔压(pulmonary artery wedge pressure,PAWP)、心输出量、混合静脉血氧饱和度(mixed venous oxygen saturation, SvO2)和PVR等,有助于判断有无心内左向右分流、评价对肺血管扩张剂的反应性和制定治疗策略。
规范操作的RHC,采集的数据才可靠。RHC过程中需要注意的几个方面:(1)全程进行心电和血压监护,必要时吸氧;(2)选择合适的静脉穿刺路径;(3)测压前校准好零点(一般采用仰卧位时第4肋间隙前胸壁至床面中点作为零点校准位,代表左心房所在水平);(4)首次导管检查或有心腔内分流患者应采集腔静脉、右心各腔室、肺动脉血测定血氧饱和度;(5)记录腔静脉、右心各腔室、肺动脉压力;(6)漂浮导管测定PAWP;(7)导管所获压力值均须在呼气末采集;(8)心输出量可以采用热稀释法测定(一般不用于有心内及大动脉水平分流患者),也可以用Fick法测得。
急性血管反应试验的目的是筛选出对口服高剂量钙通道阻滞剂(calcium channel blockers,CCBs)有效的患者。对IPAH、DPAH和HPAH患者应进行急性血管反应试验,阳性患者预后优于阴性患者。用于急性血管反应试验的药物包括吸入NO、吸入伊洛前列素、静脉用前列环素(依前列醇)和静脉用腺苷,具体用法见表8。静脉用腺苷患者耐受性差,已很少采用。
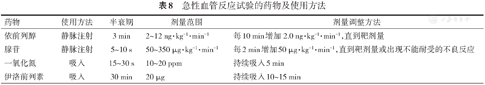
急性血管反应试验的药物及使用方法
急性血管反应试验的药物及使用方法
| 药物 | 使用方法 | 半衰期 | 剂量范围 | 剂量调整方法 |
|---|---|---|---|---|
| 依前列醇 | 静脉注射 | 3 min | 2~12 ng·kg-1·min-1 | 每10 min增加 2.0 ng·kg-1·min-1,直到靶剂量 |
| 腺苷 | 静脉注射 | 5~10 s | 50~350 μg·kg-1·min-1 | 每2 min增加50 μg·kg-1·min-1,直到靶剂量或出现不能耐受的不良反应 |
| 一氧化氮 | 吸入 | 15~30 s | 10~20 ppm | 持续吸入5 min |
| 伊洛前列素 | 吸入 | 30 min | 20 μg | 持续吸入10~15 min |
急性血管反应试验阳性标准为:用药后mPAP下降幅度≥10 mmHg,且mPAP值下降到≤40 mmHg,同时心输出量增加或不变。通常仅有10% IPAH患者可达到阳性标准。
(十二)基因检测
对PAH患者进行基因检测具有重要意义。遗传学诊断有助于PAH家系成员明确自身是否携带致病突变基因及其临床意义。携带突变基因但尚无临床表现的家族成员需要进行早期筛查并密切随访。建议筛查的PAH相关基因及高危人群见表9。

建议筛查的动脉性肺动脉高压(PAH)相关基因及高危人群
建议筛查的动脉性肺动脉高压(PAH)相关基因及高危人群
| 目的基因 | 筛查人群 | 筛查目的 |
|---|---|---|
| BMPR2 | 遗传性PAH患者及亲属 | 了解BMPR2携带情况,早期筛查无症状携带者并密切随访 |
| IPAH患者 | 了解BMPR2携带情况,帮助判断预后及制定治疗方案 | |
| ACVRL1、Endoglin、 SMAD9、BMPR1B、TBX4、CAV1、KCNK3、BMP9 | 遗传性PAH患者、IPAH患者 | 了解PAH患者致病基因携带情况 |
| Endoglin、ACVRL1 | HHT患者及其亲属 | 了解HHT的遗传信息,查找携带致病基因的家庭成员 |
| EIF2AK4 | 疑诊PVOD/PCH的患者、PVOD/PCH患者父母及子女 | 明确PVOD/PCH诊断;早期筛查无症状携带者并密切随访 |
| PTGIS | IPAH患者 | 查找合并该基因突变者,选择对伊洛前列素治疗敏感者 |
注:IPAH:特发性肺动脉高压;HHT:遗传性出血性毛细血管扩张症;PVOD/PCH:肺静脉闭塞病/肺毛细血管瘤病
PH的诊断建议从疑诊(临床及超声心动图筛查)、确诊(血流动力学诊断)、求因(病因诊断)及功能评价(严重程度评估)四个方面进行。这四个方面并非严格按照流程分步进行,临床操作过程中可能会有交叉,其中病因诊断贯穿于PH诊断的全过程。诊断策略及流程见图2。
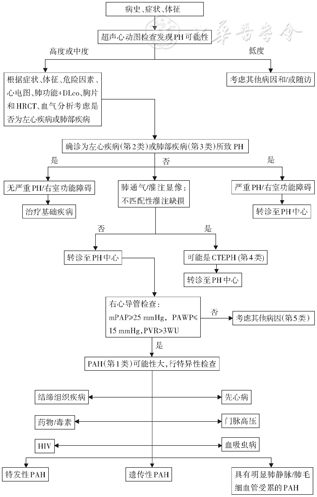
注:CTEPH:慢性血栓栓塞性肺动脉高压:DLco:CO弥散量;HIV:人免疫缺陷病毒;HRCT:高分辨率CT;mPAP:肺动脉平均压; PAH:动脉性肺动脉高压;PAWP:肺动脉楔压;PH:肺动脉高压;PVR:肺血管阻力;1 mmHg=0.133 kPa
通过病史、症状、体征以及心电图、X线胸片等疑诊PH的患者,进行超声心动图的筛查,以明确发生PH的可能性。要重视PH的早期诊断,对存在PAH相关疾病和/或危险因素,如家族史、CTD、CHD、HIV感染、门脉高压或能诱发PAH的药物或毒物摄入史者,应注意定期进行PH的筛查。
对于存在PAH相关疾病和/或危险因素的患者,如果超声心动图高度怀疑PH,需要做RHC进行诊断与鉴别诊断。
对于左心疾病或肺部疾病患者,当合并重度PH和/或右心室功能不全时,应转诊到PH中心,进一步寻找导致PH的病因。如果V/Q显像显示呈肺段分布、与通气不匹配的灌注缺损,需要考虑CTEPH。根据CTPA、RHC和肺动脉造影进行最终诊断。
对于明确诊断为PAH患者,需要根据WHO功能分级、6分钟步行试验(6 minutes walking test,6MWT)及相关检查结果等进行严重程度评估,以利于制定治疗方案。
【推荐意见】
1.推荐超声心动图作为疑诊PH患者首选的无创性检查(1C)。
2.推荐V/Q显像用于原因不明的PH患者筛查CTEPH(1C)。
3.推荐RHC作为疑诊PAH或CTEPH的确诊检查(1C)。
4.建议对IPAH、HPAH、HHT及已知基因突变患者的第一代无症状家庭成员进行遗传咨询(2C)。建议对存在基因突变的个体进行PH的相关评估(如超声心动图、心肺运动试验等)(2C)。
【推荐意见说明】
随着疾病的发展和认识的深入,很多PH患者可能会出现多种因素合并存在或相继出现,因此,在临床实践中应该对患者进行动态全面评估。
PAH是指肺动脉(主要是肺小动脉)病变所引起的肺血管阻力和肺动脉压力升高,而PAWP正常,其血流动力学定义为mPAP≥25 mmHg,PAWP≤15 mmHg和PVR>3 WU(1 WU=80 dyn·s·cm-5),主要包括IPAH、HPAH、药物或毒物相关PAH以及疾病相关性PAH[2]。
PAH的症状缺乏特异性,主要表现为劳力性呼吸困难、乏力、胸痛、晕厥以及进行性右心衰竭症状和体征[116],疾病相关性PAH还存在基础疾病的临床表现,其中晕厥、右心衰竭和疾病进展是影响预后的重要临床特征[116]。
PAH临床严重性评估是指根据临床表现、WHO功能分级、6MWT、心肺运动试验、超声心动图、CMR、血流动力学以及血清生物学标志物等多项检查指标,对患者的病情及预后进行综合评价。临床严重性评估有助于指导治疗,评估疗效。
包括WHO功能分级、6MWT和心肺运动试验等。
1.WHO功能分级:是PAH患者初诊时评估病情严重程度和预测生存的重要指标[117, 118],而治疗前后的功能分级变化也是评估疗效的主要指标。WHO功能分级(表10)用于评价PAH患者功能状态,分为Ⅰ~Ⅳ级,其分级原则与纽约心脏病协会心功能分级相似,但增加了晕厥症状的描述,Ⅰ/Ⅱ级患者的生存期显著高于Ⅲ级或Ⅳ级者,功能分级恶化是疾病进展的重要警示指标[63]。

WHO功能分级
WHO功能分级
| 分级 | 分级标准 |
|---|---|
| Ⅰ级 | 患者体力活动不受限,日常体力活动不会导致呼吸困难、乏力、胸痛或接近晕厥 |
| Ⅱ级 | 患者体力活动轻度受限,休息时无不适,但日常活动会出现呼吸困难、乏力、胸痛或接近晕厥 |
| Ⅲ级 | 患者体力活动明显受限,休息时无不适,但低于日常活动会出现呼吸困难、乏力、胸痛或接近晕厥 |
| Ⅳ级 | 患者不能进行任何体力活动。存在右心衰竭征象,休息时可出现呼吸困难和(或)乏力,任何体力活动均可加重症状 |
2. 6MWT:是测试PAH患者运动耐量以及评价疗效的重要客观检查方法,操作简便易行,首次住院PAH患者6分钟步行距离(6 minutes walking distance,6MWD)与预后密切相关[119, 120]。6MWD结果受多种因素影响,包括性别、年龄、身高、体重、并发症、需氧量、认知水平、积极性等,建议在6MWT结束时采用Borg呼吸困难评分来评价PAH患者的心肺功能和尽力程度[121]。
3.心肺运动试验:是一项客观、定量地评价心肺储备功能、运动耐量的重要检查项目,可以用于评估PAH患者的运动耐量、治疗效果和判断预后[122, 123]。心肺运动检查应该具备良好的质控[2],目前心肺运动试验大多使用逐级递增操作流程[124]。PAH患者运动耐量、有氧代谢能力和通气效率明显受损,表现为呼气末二氧化碳分压降低,二氧化碳通气量(ventilation/carbon dioxide output, VE/VCO2)升高,氧脉搏(VO2/HR)和峰值氧摄取量(peak oxygen uptake,PeakVO2)降低[125]。PAH 患者最大摄氧量(maximal oxygen uptake,VO2max)<10.4 ml·min-1·kg-1则预示死亡率明显升高[126]。
超声心动图检查有助于评价右心功能。经胸超声心动图检查能够反映PAH病情严重程度和预后的指标主要包括:右房面积、 TAPSE 、右室面积变化分数、Tei指数、心包积液等[127, 128, 129],采用斑点追踪超声心动图技术可以提高右心功能检测的准确性,研究表明二维斑点追踪超声心动图技术测量的右室应变和应变率与PAH患者的运动耐量和危险分层相关[130, 131],右心室收缩运动不同步性是IPAH患者生存率的一个独立的预测因子[132, 133, 134],三维超声心动图测量的右室游离壁应变、右室容量和右室射血分数可用于预测PAH患者的危险分层[135]。超声心动图评估右心功能的准确性不够,动态观察相关指标的变化临床意义更大。
CMR评价PAH严重程度及预后的指标包括:右室射血分数、右室搏出量、右室舒张末期容积、左室舒张末期容积、心室质量指数、主肺动脉面积变化、室间隔偏移程度、平均肺动脉血流速度及延迟强化等[136, 137, 138]。CMR对右心室的形态和功能的评估比超声心动图准确,还可以评估心输出量和每搏量,能够反映PAH严重程度及预后的CMR指标包括右心室容量增加、左心室容量降低、右心室射血分数以及每搏量降低。
PAH治疗前进行危险分层评估病情严重程度,有助于制定个体化起始治疗方案,随访中进行危险分层旨在评估治疗效果和调整治疗方案。针对PAH危险分层和预后评估进行过一些相关研究[56, 117, 144, 145, 146],但这些研究表明尚无单个指标能准确判断患者病情、评估预后和充当治疗目标,需要综合多个临床指标进行评估,根据这些指标建立了危险分层模型或评分量表。但这些模型或评分量表包含指标众多,有些并不是所有医院都能开展,有些是随访中不常规复查的指标(如心肺运动试验),因而不便于临床医师的操作实施。在2018年WSPH大会上发布了以2015年ESC/ERS PH指南中的危险分层量表为基础的简化版危险分层量表(表11)[147, 148]。
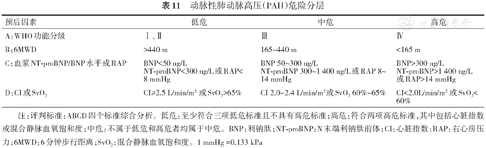
动脉性肺动脉高压(PAH)危险分层
动脉性肺动脉高压(PAH)危险分层
| 预后因素 | 低危 | 中危 | 高危 |
|---|---|---|---|
| A:WHO功能分级 | Ⅰ、Ⅱ | Ⅲ | Ⅳ |
| B:6MWD | >440 m | 165~440 m | <165 m |
| C:血浆NT-proBNP/BNP水平或RAP | BNP<50 ng/L NT-proBNP<300 ng/L或RAP<8 mmHg | BNP 50~300 ng/L NT-proBNP 300~1 400 ng/L或RAP 8~14 mmHg | BNP>300 ng/L NT-proBNP>1 400 ng/L 或RAP>14 mmHg |
| D:CI或SvO2 | CI≥2.5 L/min/m2 或SvO2>65% | CI 2.0~2.4 L/min/m2或SvO2 60%~65% | CI<2.0L/min/m2 或SvO2<60% |
注:评判标准:ABCD四个标准综合分析。低危:至少符合三项低危标准且不具有高危标准;高危:符合两项高危标准,其中包括心脏指数或混合静脉血氧饱和度;中危:不属于低危和高危者均属于中危。BNP:利钠肽;NT-proBNP:N末端利钠肽前体;CI:心脏指数;RAP:右心房压力;6MWD:6分钟步行距离;SvO2:混合静脉血氧饱和度。1 mmHg =0.133 kPa
简化版危险分层量表根据PAH患者1年预期死亡率将患者分为低危、中危或高危,低危患者1年预期死亡率<5%,中危为5%~10%,高危>10%。危险分层主要依据WHO功能分级、6MWD、生物标志物或右房压及心脏指数或SvO2等指标,具有至少3个低危指标且不具有高危指标定义为低危状态,具有至少2项高危指标(其中包括心脏指数或SvO2)定义为高危状态,不符合低危和高危者都属于中危状态。简化版的危险分层通过对低、中、高危进行详细的定义,使危险分层更加明确、便于临床应用。
【推荐意见】
1.推荐综合多项指标对PAH患者病情严重程度进行评估,包括临床特征、运动耐量、生物标志物、超声心动图及血流动力学指标等(1C)。
2.推荐病情稳定的PAH患者每3~6 个月随访评估1次(1C)。
【推荐意见说明】
以往针对临床严重程度评估和危险分层的研究多数纳入的是成人PAH患者,因此这种危险分层是否适合其他类型PH尚不清楚。
1.体力活动和专业指导下的康复:PAH患者应在药物治疗的基础上、在专业指导下进行运动康复训练。最近两项随机对照研究表明每周进行低负荷的运动康复可以改善患者6MWD、PeakVO2以及血流动力学参数[149, 150]。一项荟萃分析纳入16项研究,患者469例,分析发现运动康复可以改善PAH患者6MWD、心肺功能和生活质量评分[151]。
2.妊娠、避孕及绝经后激素治疗:随着靶向药物的广泛应用,妊娠PAH患者死亡率有所下降,但仍在5%~23%,且妊娠并发症多,因此,建议PAH患者避免怀孕。若妊娠期间被确诊为PAH,最好在孕22周前终止妊娠;选择继续妊娠者,必须转至专业的PH中心进行全面评估和密切随访。可以考虑给予前列环素类似物或磷酸二酯酶5型(phosphodiesterase type 5,PDE5)抑制剂治疗,尽量降低肺动脉压及PVR[152, 153, 154]。
3.择期手术:对PAH患者即使进行择期手术也会增加患者风险,接受择期手术者,硬膜外麻醉可能比全身麻醉耐受性好。
4.预防感染:PAH患者容易合并肺部感染,而肺部感染是加重心衰甚至导致死亡的重要原因之一。因此,尽管没有临床对照试验证据,仍推荐PAH患者预防性应用流感疫苗和肺炎链球菌疫苗[155, 156]。
5.社会心理支持:研究显示PAH对患者情绪产生重大影响,焦虑发生率20%~40%,抑郁发生率21%~55%,58%的PAH患者存在认知后遗症[157, 158]。因此,应充分评估患者的精神心理状态,鼓励家属给予心理支持,必要时请专科进行干预和支持。
6.旅行:对于WHO功能分级为Ⅲ~Ⅳ级、动脉血氧分压低于60 mmHg的PAH患者,在航空旅行时建议吸氧。PAH患者应避免前往海拔高于1 500~2 000 m以上地区。
7.遗传咨询:对筛查出基因突变的患者,需要告知关于遗传学变异的可能性,以及家庭成员可能携带相关的突变致PAH的风险增加,并建议相关家庭成员进行筛查及早期诊断。基因检测和遗传学咨询严格遵循当地法规的要求,遵循伦理原则。
【推荐意见】
1.推荐PAH患者在专业医生的指导下,在药物治疗的基础上进行运动康复训练(1A)。
2.推荐育龄期女性PAH患者避孕(1C)。
3.PAH患者进行择期手术时,建议首选局部麻醉或硬膜外麻醉,尽量避免全身麻醉(2C)。
4.推荐PAH患者接受流感病毒或肺炎疫苗注射以预防感染(1C)。
5.推荐给予PAH患者社会心理支持(1C)。
1.抗凝治疗:早期对IPAH患者进行尸检发现半数以上存在血栓形成,抗凝治疗与预后改善相关[159];此后荟萃分析显示华法林抗凝治疗能改善IPAH患者预后[160];但这些研究大多是在靶向治疗时代前的回顾性研究,缺乏随机对照研究数据。近年PAH注册登记研究和系统性回顾分析显示抗凝治疗存在不一样的效果[161],抗凝治疗对SSc-PAH患者不能获益甚至会增加死亡风险[162]。
2.利尿剂:PAH患者出现失代偿性右心衰竭时导致液体潴留、中心静脉压升高、肝瘀血、多浆膜腔积液等,利尿剂可改善上述状况,但目前尚没有应用利尿剂的随机对照研究。常用利尿剂包括袢利尿剂(呋塞米、托拉塞米)和醛固酮受体抑制剂(安体舒通)。近年排水型利尿剂血管加压素V2受体拮抗剂(托伐普坦)也尝试在这类患者中应用,不降低有效循环血容量,但确切疗效及安全性尚需经进一步临床研究证实。应用利尿剂治疗时需要监测体重、肾功能、电解质等血生化指标,避免低血容量和电解质紊乱。
3.氧疗:目前尚缺乏随机对照研究证实PAH患者长期氧疗获益。基于COPD患者的证据,建议动脉血氧分压低于60 mmHg(外周血氧饱和度<91%)的PAH患者进行氧疗,以使动脉血氧分压≥60 mmHg(外周血氧饱和度≥91%)。
4.地高辛及其他心血管药物:地高辛可以增加心脏收缩力,改善心输出量,但其在PAH患者中的长期疗效尚不确切;可用于降低PAH患者发生快速房性心律失常的心室率。不建议应用血管紧张素转化酶抑制剂、血管紧张素Ⅱ受体拮抗剂、β受体阻滞剂、硝酸酯类或伊伐布雷定等药物治疗PAH,如因合并左心疾病(高血压、冠心病等)需要应用以上药物者,需观察血压、心率等,注意药物间相互作用。
5.贫血的治疗:研究显示PAH包括IPAH、CHD-PAH以及CTD-PAH等患者常伴有铁缺乏,并且铁缺乏与PAH严重程度和预后相关[163, 164]。一项开放性非随机对照临床试验结果显示[165],缺铁的 PAH 患者经静脉补铁治疗 2 个月后缺铁状况和6MWD明显改善。另一项多中心、随机、双盲、安慰剂对照、交叉临床试验结果显示,静脉铁剂治疗的IPAH 患者PVR明显降低。研究显示IPAH 患者肠道对铁的吸收减少,缺铁的患者接受口服铁剂治疗 4 周后仅有44%患者铁蛋白轻度升高[164]。
【推荐意见】
1.建议对IPAH、HPAH和食欲抑制剂相关PAH患者进行个体化抗凝治疗(2C)。
2.推荐对存在右心功能不全、液体潴留的PAH患者进行利尿治疗(1C)。
3.不推荐PAH患者应用血管紧张素转化酶抑制剂、血管紧张素Ⅱ受体拮抗剂、β受体阻滞剂、硝酸酯类或伊伐布雷定等药物(除非合并左心疾病,如高血压、冠心病等)(1C)。
4.推荐PAH患者进行铁代谢检测(1C), 对铁缺乏的PAH患者进行补铁治疗(首选静脉补铁)(2C)。
1.CCBs:急性血管反应试验阳性患者建议给予足量CCBs治疗,心率偏慢者考虑应用硝苯地平和氨氯地平,心率偏快者倾向于应用地尔硫 。建议起始低剂量,逐渐增加至可耐受的最高剂量,硝苯地平120~240 mg/d,地尔硫
。建议起始低剂量,逐渐增加至可耐受的最高剂量,硝苯地平120~240 mg/d,地尔硫 240~720 mg/d,氨氯地平最高可达20 mg/d。
240~720 mg/d,氨氯地平最高可达20 mg/d。
未进行急性血管反应试验或者反应阴性的患者因低血压、晕厥、右心衰竭等可能的严重副作用,不应使用CCBs类药物。对于其他类型的PAH患者,急性血管反应试验无法预测CCBs的长期疗效,亦不推荐使用CCBs。
2.内皮素受体拮抗剂(endothelin receptor antagonist,ERA):内皮素在PAH发病中起重要作用。内皮素-1可通过与肺血管平滑肌细胞中的内皮素受体A和B结合,引起血管收缩,促进有丝分裂,参与PAH的发生发展。ERA可以通过干预内皮素途径治疗PAH。
波生坦:波生坦是第一个合成的ERA类药物,为内皮素受体A、B双重拮抗剂。波生坦可以改善IPAH、CTD-PAH、CHD-PAH、HIV-PAH患者运动耐量、心功能分级、血流动力学参数以及临床恶化时间[166, 167, 168, 169],延展研究显示波生坦治疗组3年存活率好于传统治疗。
安立生坦:安立生坦是高选择性内皮素A受体拮抗剂。研究显示安立生坦5 mg和10 mg两个剂量均能显著改善患者6MWD,呈较明显的剂量-效应关系[170, 171]。对PDE5抑制剂治疗反应不理想的PAH患者序贯联合安立生坦治疗24周,能明显改善患者运动耐量和血流动力学参数[172]。与单药治疗相比,初始联合安立生坦和他达拉非可明显降低临床恶化事件发生率,这种起始联合方案对于WHO 功能分级Ⅱ级的患者中的获益不亚于Ⅲ级的患者[173, 174]。国人研究也显示安立生坦能改善PAH患者12周运动耐量、心功能等[175],序贯联合他达拉非治疗能改善运动耐量,降低临床恶化事件发生率[176]。
马昔腾坦:马昔腾坦是新一代双重ERA,具有更好的组织穿透力和受体亲和力。一项随机对照研究显示,与安慰剂相比马昔腾坦10 mg单药或联合治疗均能显著降低患者疾病恶化/死亡风险和因PAH导致的死亡率或住院率,改善患者6MWD、WHO功能分级、生活质量、血流动力学参数和NT-proBNP。对接受背景治疗的患者进行亚组分析[177],马昔腾坦序贯联合治疗与安慰剂组相比能明显降低患者疾病恶化/死亡风险和因PAH导致的死亡率或住院率。在新发PAH患者中初始联合马昔腾坦和他达拉非治疗的研究,结果显示16周PVR下降47%、6MWD,WHO功能分级和NT-proBNP也有获益[178]。
3.PDE5抑制剂:NO是重要的血管扩张因子,通过维持血管平滑肌细胞内环磷酸鸟苷(cyclic guanosine monophosphate,cGMP)浓度到达扩血管效应。肺血管包含大量的PDE5,它是cGMP的降解酶。PDE5抑制剂可以通过减少cGMP的降解,升高其浓度引起血管舒张。此外,PDE5抑制剂还有抗增殖的作用。
西地那非:西地那非是一种特异性PDE5 抑制剂。一项纳入278 例PAH患者的随机对照研究显示[179],西地那非明显改善患者6MWD、WHO功能分级以及血流动力学;对西地那非单药治疗的患者随访3年,6MWD和WHO功能分级的改善得以维持[180]。国内研究显示西地那非能改善中国PAH患者运动耐量及血流动力学[181, 182];与传统治疗相比,西地那非可改善PAH患者1年、2年和3年的生存率[183, 184]。
他达拉非:他达拉非是一种长效的PDE5抑制剂。一项纳入405例PAH患者,随机给予安慰剂及他达拉非2.5、10、20或40 mg治疗16周,结果显示40 mg组能明显改善PAH患者6MWD、WHO功能分级和临床恶化出现的时间[185]。波生坦序贯联合他达拉非较单用波生坦组6MWD也明显改善。对接受他达拉非20或40 mg的PAH患者进行52周随访[186],结果发现6MWD得以继续维持。
伐地那非:伐地那非是一种高选择性PDE5抑制剂。一项在中国PAH患者进行的随机双盲安慰剂对照研究[187],66例患者随机分为伐地那非组与安慰剂对照组,主要终点为6MWD,结果显示伐地那非能明显改善中国PAH患者运动耐量。伐地那非的不良反应与西地那非类似。
4.可溶性鸟苷酸环化酶(soluable guanylate cyclase,sGC)激动剂:利奥西呱是一种新型的sGC激动剂,具有独特的双重激活sGC机制,其作用效果不依赖于体内NO水平,可单独或与NO协同提高血浆中的cGMP水平,引起血管舒张和抗重塑作用。一项研究纳入443例PAH患者[188],其中50%的患者接受过背景治疗。结果显示,与安慰剂相比,利奥西呱能明显改善PAH患者运动耐量、血流动力学、心功能分级,降低NT-proBNP水平,降低临床恶化事件发生率。对其中入选的77例中国患者进行分析显示疗效和安全性与总体一致。
利奥西呱联合西地那非在PH患者中的安全性和有效性研究[189],结果显示两药联合组低血压发生率明显升高,而血液动力学参数或运动能力无明显差异,因此,不建议PDE5抑制剂和利奥西呱联合使用。
一项前瞻性开放标签多中心的单臂研究中[190],61例PDE5抑制剂治疗反应不足的PAH患者换用利奥西呱治疗,24周时患者6MWD增加、WHO 功能分级改善、NT-proBNP降低,研究显示治疗反应不足的PAH患者可能从PDE5抑制剂转换为利奥西呱的治疗中获益。
5.前列环素类似物和前列环素受体激动剂:前列环素由血管内皮细胞产生,具有强效扩张血管作用,也是目前最强的内源性血小板聚集抑制剂。研究表明PAH患者肺动脉中前列环素合成酶的表达下降,尿中代谢水平降低,人工合成的前列环素类似物可用于治疗PAH。
依前列醇:依前列醇是第一个人工合成的前列环素类似物,半衰期短(3~5 min),需要持续深静脉注射给药。长期观察表明静脉注射依前列醇能改善心功能Ⅲ-Ⅳ级的IPAH患者的生存率[191, 192],并且功能分级、运动耐量和血流动力学均获得明显改善。此外,研究也报道静脉注射依前列醇对于疾病相关PAH也具有同样疗效[193]。
伊洛前列素:伊洛前列素是一种前列环素类化合物,可通过肺泡型雾化装置给药,研究显示WHO功能Ⅲ/Ⅳ级PAH和不宜手术的CTEPH患者[194],雾化吸入伊洛前列素(30 μg/d)与安慰剂相比,能明显改善6MWD和WHO功能分级;长期研究结果显示伊洛前列素能改善PAH患者运动耐量、血流动力学以及生存率[195, 196]。国内研究显示常规半剂量(15 μg/日)的伊洛前列素雾化吸入也能明显改善PAH患者运动耐量和功能分级[197];伊洛前列素起效迅速,20 μg雾化吸入可以作为PAH患者急性肺血管反应试验的药物并具有很好的耐受性[198]。
曲前列尼尔:曲前列尼尔在室温下化学性质稳定,半衰期长(2~4 h),与依前列醇具有相似的药理学性质。多项临床研究证实曲前列尼尔长期应用的有效性和安全性。曲前列尼尔单药治疗1年和4年的生存率分别是88%和70%[199]。皮下注射曲前列尼尔最常见不良反应为注射部位疼痛,需在有经验的中心指导局部注射部位的护理。静脉注射曲前列尼尔也显示了其短期和长期疗效。一项为期12周的随机、双盲临床研究显示[200],与安慰剂组比,持续静脉注射曲前列尼尔(平均剂量72 ng·kg-1·min-1)明显改善6MWD、NYHA(纽约心脏协会)心功能分级。
司来帕格:司来帕格是一种长效的口服前列环素受体激动剂。一项以事件驱动为终点的Ⅲ期临床试验[201],纳入1 156例PAH患者,结果显示与安慰剂相比,不管是否接受背景治疗,司来帕格使PAH患者恶化/死亡事件的风险显著降低40%,包括6MWD,WHO功能分级等次要终点均明显改善。在该研究中[202],80%的患者接受背景治疗,其中15%为ERA,32%为PDE5抑制剂,33%为ERA+PDE5抑制剂,结果显示序贯联合司来帕格,可使恶化/死亡终点风险下降43%,与总体结果一致。
1.靶向药物联合治疗:PAH是一个进展性疾病,延迟达标治疗(达到低危状态)可能会影响患者的长期预后。建议PAH起始联合治疗,尽早达标。对于初治PAH患者,若为低或中危状态,起始联合不同通路靶向药物治疗(表12),若为高危状态起始联合应包括静脉前列环素类靶向药物治疗。

靶向药物用法及常见不良反应
靶向药物用法及常见不良反应
| 靶向药物 | 用法 | 不良反应 |
|---|---|---|
| 前列环素类似物 | ||
依前列醇 | 2~4 ng·kg-1·min-1起始持续静脉泵入,逐渐加到目标剂量 | 头痛、消化道症状、输注路径感染 |
伊洛前列素 | 每次10~20 μg,吸入6~9次/d | 头痛、脸红、低血压 |
曲前列尼尔 | 1.25 ng·kg-1·min-1起始,静脉或皮下注射,逐渐加到目标剂量 | 输注部位疼痛、头痛、腹泻 |
贝前列素 | 20~80 μg,qid,口服 | 头痛、面色潮红 |
| 前列环素受体激动剂 | ||
司来帕格 | 200 μg,bid 逐渐上调至耐受剂量,最大剂量1 600 μg bid | 头痛、腹泻、恶性呕吐、下颌疼痛 |
| 内皮素受体拮抗剂 | ||
波生坦 | 62.5~125 mg,bid | 转氨酶升高、外周水肿、贫血 |
安立生坦 | 5~10 mg, qd | 头痛、外周水肿、贫血 |
马昔腾坦 | 10 mg, qd | 贫血 |
| 磷酸二酯酶5型抑制剂 | ||
西地那非 | 20 mg,tid | 头痛、脸红、视觉障碍等 |
他达那非 | 20~40 mg,qd | 头痛、脸红、肌痛 |
伐地那非 | 5 mg,bid | 头痛、脸红、肌痛 |
| 鸟苷酸环化酶激动剂 | ||
利奥西呱 | 1 mg,tid,根据血压情况每2周上调一次剂量,直至2.5 mg,tid | 消化道症状、低血压、咯血 |
注:qd:1次/d;bid:2次/d;tid:3次/d;qid:4次/d
对于经治PAH患者,若仍未达到低危状态,需进行序贯联合治疗。已有多项临床研究证实序贯联合较单药治疗能取得更好疗效[188, 203]。近年来以临床事件驱动的随机对照研究显示,双药或三药序贯联合治疗组的死亡/住院风险较对照组明显降低。国内一项随机对照研究入选124例接受安立生坦治疗至少4个月的PAH患者,序贯联合他达拉非治疗16周,较单药安立生坦组6MWD明显改善,临床恶化事件发生率显著降低[176]。一项纳入4 095例PAH患者的荟萃分析显示,序贯联合治疗能将临床恶化风险降低达35%[204]。
2.药物相互作用:靶向药物联合治疗时需考虑到药物间的相互作用。波生坦是细胞色素P450同工酶CYP2C9和CYP3A4的诱导物,当通过该同工酶代谢的药物与波生坦同时应用时,其浓度就会降低,而抑制这些酶可提高波生坦血药浓度。西地那非由细胞色素P450同工酶CYP3A4(主要途径)和CYP2C9(次要途径)代谢,当存在CYP3A4底物、抑制剂及CYP3A4底物联合β受体阻滞剂时,西地那非生物利用度升高、清除率降低。当西地那非与波生坦等P450同工酶诱导物合用时会导致清除增加,从而影响西地那非疗效,因此,临床中合并用药时需要注意。为避免体循环低血压,当PAH靶向治疗联合应用抗高血压药物时需要谨慎,例如β受体阻滞剂、血管紧张素转换酶抑制剂等。
【推荐意见】
1.对于IPAH、HPAH、DPAH患者,若急性血管反应试验阳性,推荐使用能够耐受的高剂量CCBs(1C),推荐进行密切随访,3~6个月评价患者功能状态及血流动力学指标(1C);若未行急性血管反应试验或结果阴性,不建议使用CCBs治疗(2C)。
2.推荐PAH在病情早期(低危或中危)进行靶向药物联合治疗(1B)。
3.推荐高危PAH患者靶向药物联合治疗,方案中应包括静脉前列环素类似物(1A)。
【推荐意见说明】
应用高剂量CCBs后WHO功能分级Ⅰ、Ⅱ级且血流动力学明显改善(接近正常)的IPAH、HPAH、DPAH患者推荐继续应用高剂量CCBs治疗。应用高剂量CCBs后WHO功能分级或血流动力学无显著改善(未接近正常)的患者,推荐逐渐减量至停用CCBs,并开始应用靶向治疗。
通过BAS建立心房内右向左分流可以降低右心的压力,增加左心室前负荷和心输出量。BAS的实施尽管降低了动脉血氧饱和度,但可改善体循环氧气的转运,同时可降低交感神经过度兴奋[205]。一项纳入204例PAH的荟萃分析显示,BAS术后右房压明显降低,心输出量增加,术后48 h内、30 d内和>30 d死亡率分别为4.8%、14.6%和37.7%[206]。报道显示BAS使患者等待肺移植术的成功率为30%~40%[207]。建议BAS可作为经充分内科治疗效果不佳等待肺移植的桥接治疗。BAS作为姑息治疗或桥接治疗方法,建议在有经验的中心进行。操作前细致的风险评估可降低死亡率,终末期患者右房平均压>20 mmHg且在呼吸空气静息状态下氧饱和度<85%时,不能行BAS。
肺移植和心肺联合移植治疗PH开始于80年代初[208]。对于治疗无效或WHO功能分级维持在Ⅲ级或Ⅳ级的PAH患者建议行肺移植[191, 192, 209]。国外研究提示:IPAH占肺移植受者的2.9%,其他类型PH占1.6%[210]。
PAH肺移植术后5年生存率为45%~50%,生活质量明显提高[210, 211]。因此当初始联合治疗仍然疗效不佳时应尽快进行肺移植术前评估。PVOD和PCH缺乏有效治疗药物,应在诊断同时进行肺移植评估。
PH患者移植评估标准和移植标准,多采用2014年国际心肺移植协会更新的标准[212]。
1.移植评估标准:(1)充分内科治疗后仍为WHO功能分级Ⅲ或Ⅳ级;(2)疾病进展迅速;(3)需使用静脉前列环素类似物治疗;(4)已知或可疑PVOD或PCH。
2.移植标准:(1)包括前列环素类似物在内的药物联合治疗至少3个月,仍为WHO功能分级 Ⅲ或Ⅳ级;(2)心脏指数<2 L/(min·m2);(3)右房压>15 mmHg;(4)6MWD<350 m;(5)出现明显咯血、心包积液或进行性右心衰竭的征象(如肾功能不全、胆红素升高、BNP或NT-proBNP升高等)。
目前国际心肺移植协会对于绝大部分PAH患者推荐双肺移植。由简单分流性先天性心脏病引起的艾森曼格综合征可选择双肺移植+心脏缺损修补术或心肺联合移植[211],室间隔缺损引起的PAH行双肺移植+心脏缺损修补术更能获益[213]。进入等待肺移植名单的受者建议术前进行康复训练,并可从中获益[214]。
PAH肺移植在过去的数年中进步很大,其中最重要的就是术中和术后体外膜肺氧合(extracorporeal membrane oxygenation,ECMO)的应用[215]。术中ECMO基本上取代了传统的体外循环,可减少肾功能衰竭等围术期并发症、减少血制品的应用,患者可获得更好的生存[216, 217]。在某些PAH和右心功能衰竭的肺移植受者,于手术麻醉前置入外周静脉-动脉ECMO(V-A ECMO)可避免血流动力学不稳定。延长ECMO支持时间可预防肺移植术后早期移植物失功。由于PAH肺移植的数量少、风险高,因此建议在有经验的肺移植中心进行。
【推荐意见】
对于包括前列环素类药物在内的靶向药物联合治疗至少3个月后、危险分层为中、高危的患者,建议进行肺移植评估(2C)。
【推荐意见说明】
在启动肺移植评估前反复进行风险评估很重要。建立风险评估工具,根据本指南推荐的风险评估策略决定患者去肺移植中心咨询的时机。尽管重症监护病房(intensive care unit,ICU)维护水平和高级生命支持技术有进步,但理想的受者仍是康复状态良好的患者。
合并有其他疾病(包括大手术)和(或)存在右心衰竭的患者需要在ICU管理。有资料显示,ICU的PAH患者病死率高达41%,预后差,因此重症PAH患者应在PH中心的ICU管理。基本监测指标有生命体征(心率、血压、体温、血氧饱和度)、出入量、中心静脉压、中心静脉血氧饱和度和血乳酸含量,中心静脉血氧饱和度低于60%合并乳酸水平升高,少尿或无尿均提示右心衰竭,在某些情况下,还需放置漂浮导管进行全面血流动力学监测。
重症PAH右心衰竭的患者常因相关诱因而诱发,包括感染、贫血、甲状腺功能障碍、肺栓塞、心律失常或不遵医嘱服药。室上性快速心律失常,尤其心房扑动和心房颤动,是重症PAH患者右心衰竭的常见诱因,需尝试快速恢复窦性心律。对于重症患者,容量管理极为关键。大多数患者右心室充盈压明显升高,心输出量下降,补液可进一步增加右心室充盈压力和心室容积,从而加剧室间隔向左侧移位并增加三尖瓣反流[218],导致左心室充盈下降和功能进一步恶化,对于这些患者应静脉注射袢利尿剂甚至血液滤过以寻求容量负平衡。
低心输出量患者可能需要使用正性肌力药,多巴酚丁胺是最常用的正性肌力药物。血压低的患者可能需要升压治疗维持各脏器灌注,去甲肾上腺素是首选的升压药物,可避免心率增快以及左心室充盈减少致心输出量进一步下降和左室舒张末压力升高。PH动物模型显示,米力农和左西孟旦可改善右心室功能,降低PVR,个案报道可用于PAH合并右心衰竭患者[219]。常用的血管活性药物见表13[209]。

临床用于治疗重症右心衰竭的血管活性药物
临床用于治疗重症右心衰竭的血管活性药物
| 药物 | 心排量 | PVR | SVR | 心律失常 |
|---|---|---|---|---|
| 多巴酚丁胺 | ||||
<5 μg·kg-1·min-1 | ↑ | ↓ | ➝或↓ | ++ |
5~15 μg·kg-1·min-1 | ↑↑ | ➝ | ↓ | +++ |
| 多巴胺 | ||||
2.5~5 μg·kg-1·min-1 | ↑ | ? | ↑↑ | |
>5 μg·kg-1·min-1 | ↑ | ↑ | +++ | |
| 米力农 | ↑↑ | ↓ | ↓↓ | +++ |
| 左西孟旦 | ↑↑ | ↓ | ↓↓ | + |
| 去甲肾上腺素 | ↑ | ➝或↑ | ↑↑ | ++ |
注:PVR:肺血管阻力;SVR:全身血管阻力
降低右心室后负荷:靶向药物可用于PAH伴发右心衰竭的患者,以降低右室后负荷。静脉注射前列环素类似物起效快,常作为首选。起始三联治疗(静脉注射前列环素类似物、PDE5抑制剂和ERA)对于新诊断的PAH合并右心衰竭患者具有良好的短期和中期效果[220]。
研究显示V-A ECMO和插入肺动脉和肺静脉或左房之间的无泵型ECMO,可用于重症PAH过渡到肺移植或恢复[221, 222]。孤立性右室辅助装置(isolated right ventricular assist device,RVAD)在PAH的应用仅有个别报道[221],尚无长期成功使用的报道。
为避免右心衰竭患者全身麻醉和气管插管相关的风险和并发症,并且防止机械通气产生的负面影响,如呼吸机相关性肺炎,ECMO优先用于清醒、能自主呼吸的患者。有证据显示清醒状态下的ECMO策略是可行的,能维持长达数周的桥接时间,比包括插管和机械通气在内的桥接策略结局更好[223, 224]。应避免在尚未接受肺移植评估的患者中使用ECMO,除非存在恢复的可能,如既往病情稳定,存在右心衰竭可逆病因(如心律失常或感染)的患者,或迄今未经治疗或新诊断PAH药物治疗尚不充分的患者。
【推荐意见】
1.推荐重症PAH患者在PAH中心接受重症监护治疗(1B)。
2.建议重症PAH患者接受V-A ECMO辅助和肺动脉-左房之间的无泵型ECMO过渡到肺移植或恢复(2C)。
在PH中心确诊的PAH初始治疗患者,建议接受一般治疗及支持治疗。对于IPAH、HPAH和DPAH患者进行急性血管反应试验,阳性者逐步滴定后给予高剂量CCBs治疗;治疗3~6个月后进行全面评估,如血流动力学持续改善,且WHO功能维持Ⅰ~Ⅱ级的患者建议继续高剂量CCBs治疗,否则应启用靶向药物治疗。
急性血管反应试验阴性的患者建议初始靶向药物联合治疗,高危的患者建议联合静脉前列环素类药物。
对以下患者可考虑初始单药治疗:(1)急性血管反应试验阳性的IPAH、HPAH和DPAH患者,在CCBs治疗1年后WHO功能分级仍为Ⅰ/Ⅱ级,且有持续的血液动力学改善(与最初急性血管反应试验结果相同或更好);(2)长期接受单药治疗(>5~10年),病情稳定于低危状态的PAH患者;(3)年龄>75岁的IPAH患者,存在多个射血分数保留左心衰竭的危险因素(高血压、糖尿病、冠心病、房颤、肥胖);(4)疑诊或高度可能是PVOD/PCH患者;(5)HIV、门脉高压或未矫正的CHD等相关PAH患者(上述患者未纳入起始联合的临床随机对照研究);(6)轻症PAH患者(如WHO功能分级Ⅰ级,PVR 3~4 WU,mPAP<30 mmHg,超声心动图提示右心室功能正常);(7)无法获得联合治疗或存在禁忌证(如严重肝病)[148]。
治疗3~6个月进行评估,若为低危状态,应继续治疗并规律随访;若为中危状态,推荐三种靶向药物联合使用;若为高危状态,建议使用包括静脉注射前列环素类药物的联合治疗方案,并进行肺移植评估。病情持续恶化患者,可考虑BAS作为姑息性或肺移植前的桥接性治疗。PAH患者的治疗流程见图3。

注:PH:肺动脉高压;PAH:动脉性肺动脉高压;CCB:钙通道阻滞剂;IPAH:特发性肺动脉高压;HPAH:遗传性肺动脉高压;DPAH:药物和毒物相关性肺动脉高压
【推荐意见】
推荐给予充分的药物治疗,使PAH患者病情达到或维持低危状态(1C)。
【推荐意见说明】
尽管不少靶向药物应用于临床,但目前这些靶向药物还很难使肺动脉压力降至正常,因此有必要对PAH患者进行危险分层,确定治疗目标。
PAH患者总体的治疗目标是达到低危状态,表现为良好的运动耐量、生活质量、右心功能和低死亡风险。为便于临床医师推广和操作,评估危险分层的指标进行了简化。建议患者每3~6个月进行随访评估,随访检查项目包括WHO功能分级、血常规、血生化、动脉血氧饱和度、BNP或NT-proBNP、6MWT、超声心动图等,根据患者病情和用药情况进行其他检查项目包括铁代谢、D-二聚体、肌钙蛋白、甲状腺功能等。建议在调整治疗方案或临床恶化时复查RHC。
【推荐意见】
1.推荐定期对PAH进行多项指标评估,包括临床特征、运动耐量、血清生物标志物、超声心动图及血流动力学指标等(1C)。
2.推荐病情稳定的PAH患者每3~6个月随访评估1次(1C)。病情不稳定建议及时复诊(2C)。
1.定义及分期:CHD-PAH是指由体-肺分流型CHD所引起的肺动脉压力升高。临床上分为艾森曼格综合征、体-肺分流相关PAH、PAH合并小缺损和术后PAH等四类(表14)[225]。CHD-PAH是我国PAH最常见的原因,诸多CHD患者因合并PAH而失去手术机会。

先天性心脏病相关肺动脉高压(CHD-PAH)的临床分类
先天性心脏病相关肺动脉高压(CHD-PAH)的临床分类
| 分类 | 说明 |
|---|---|
| 艾森曼格综合征 | 包括所有心内和心外大缺损所致的体-肺分流,逐渐进展为PVR严重升高及逆向(肺-体)或双向分流,常表现为紫绀、继发性红细胞增多症和多器官受累 |
| 体-肺分流相关PAH | 可纠正的(通过手术或介入) 不可纠正的 包括中到大缺损;PVR轻到中度升高,持续性体-肺分流,但静息状态下紫绀不明显 |
| PAH合并小缺损/合并缺损a | PVR明显升高同时合并小心脏缺损(通常指超声心动图评估的室间隔缺损直径<1 cm、房间隔缺损直径<2 cm,缺损本身并不是导致PVR升高的主要原因;其临床表现与IPAH相似,修复缺损是禁忌证 |
| 缺损修复后的PAH | 先天性心脏病纠正后,PAH在术后依然存在或术后术月至数年内复发,且无术后残余分流 |
注:a缺损大小适用于成人患者,其考虑因素包括直径、压力梯度、分流大小和方向、以及肺-体流量比;PVR:肺血管阻力;IPAH:特发性肺动脉高压
根据血流动力学特点,将CHD-PAH分为动力型和阻力型。(1)动力型PAH期:患者存在PAH,但肺血管尚未发生严重病变,关闭缺损之后肺动脉压力可降至正常。(2)阻力型PAH期:肺血管已发生不可逆病变,关闭缺损后,患者肺动脉压力不能降至正常或反而升高,出现术后持续PAH[35]。
2.诊断与评估:CHD的诊断主要依靠临床表现、胸部X线、超声心动图、心脏CT血管成像(computed tomography angiogram,CTA)以及心血管造影等。如何将动力型和阻力型PAH完全分开,目前尚无统一标准,一些临床指标可辅助判断。动力型PAH的患者以小婴儿为主,偶见于大龄儿童或成人,临床上通常无发绀,有充血性心力衰竭表现,儿童患者常有生长发育受限,心脏听诊缺损部位杂音响亮,动脉血氧饱和度正常,胸部X线则提示心影增大,肺血增多;超声心动图显示缺损部位以左向右分流为主,左心室增大。阻力型PAH患者则以大龄儿童及成人多见;常存在紫绀或有活动后紫绀,听诊缺损部位杂音减弱,肺动脉瓣听诊区第二心音亢进;胸部X线显示心影不大,中心肺动脉扩张而外周纤细,呈残根状;超声心动图显示缺损部位双向分流或右向左分流,且左心室内径缩小,晚期可有右室功能受损的表现。
RHC是判断CHD患者能否手术治疗和预后的最重要检查方法。CHD-PAH患者必须同时测量左、右心系统各腔室压力和血氧含量以及PAWP。通常采用Fick法测量心输出量,然后计算肺循环血流量/体循环血流量比值(Qp/Qs)、PVR、肺血管阻力与体循环阻力比值(Rp/Rs)等指标。PVR和Rp/Rs是衡量CHD-PAH手术指征的重要指标。对于儿童患者,建议采用PVR 指数(pulmonary vascular resistance index,PVRI)[PVRI=PVR×体表面积(WU·m2)]。
【推荐意见】
1.推荐RHC作为CHD-PAH患者确诊、分类及治疗指导的重要检查手段(1C)。
2.推荐根据临床表现、影像学检查以及血流动力学指标综合评估手术指征(1C)。
【推荐意见说明】
2015年ESC/ERS PH指南确定的CHD-PAH手术指征为PVR<2.3 WU或PVRI<4 WU·m2。若PVR在2.3~4.6 WU或PVRI 4~8 WU·m2,需要个体化判断术后PH改变是否可逆。若PVR>4.6 WU或PVRI>8 WU·m2则为手术禁忌[2]。也有儿科专家认为PVRI<6 WU·m2且Rp/Rs<0.3是良好的手术适应证,急性血管反应试验时PVRI下降幅度明显(>20%)且可达到上述标准的患者可能也适宜手术[226]。
3.治疗:应根据PAH血流动力学决定CHD-PAH治疗方案:对于动力型PAH(有矫治适应证)患者,手术关闭缺损是解决PAH的根本方法,应及早进行缺损的修补或介入封堵治疗,避免长期大量分流导致不可逆的肺血管重塑。阻力型PAH可采用靶向药物长期治疗和心肺联合移植或肺移植联合心脏缺损修补术。对于直接关闭缺损危险性大的“边缘型”PAH患者,可先给予靶向药物治疗或行封堵试验,观察血流动力学变化,然后确定治疗方案,对于CHD-PAH外科或介入治疗术后仍存在PAH患者建议应用靶向药物治疗。
随着靶向药物的应用,CHD-PAH患者的活动能力和预后得到明显改善。大量临床研究表明PDE5抑制剂、ERA和前列环素类似物、IP受体激动剂等靶向药物在患者中具有较好的安全性和有效性[227]。
存在严重肺血管病变的艾森曼格综合征患者,经过有效的药物治疗,可改善生活质量,延长寿命[228, 229]。对终末期肺血管疾病患者,心肺移植是最后的治疗手段[230, 231]。
【推荐意见】
1.建议艾森曼格综合征患者应用ERAs、PDE5抑制剂及前列环素类药物单药或联合治疗(2C)。
2.不推荐艾森曼格综合征患者应用CCBs(1C)。
【推荐意见说明】
随着靶向药物的应用,CHD-PAH患者的活动耐量和预后得到明显改善。但是,目前绝大部分的靶向药物仅限用于PAH,若患者同时合并存在第2大类PH,常禁用靶向药物。对于存在分流的CHD-PAH患者,无论急性血管反应试验阳性与否,都不建议用CCBs。
目前有国际指南采用mPAP>20 mmHg、PVR≥3 WU的血流动力学标准来诊断CHD-PAH[232],对于mPAP在21~24 mmHg的CHD患者是否按PAH给予靶向药物治疗,目前无确切的循证医学证据,需要根据患者的具体情况进行个体化治疗,并加强观察与随访。
CTD-PAH的发病机制和病理生理过程较为复杂,可涉及到各种类型PAH,甚至可出现数种类型混合并存的情况,其中以引起肺动脉病变导致PAH最常见。
1.诊断与评估:CTD是引起PAH的重要原因,而PAH也是CTD的严重并发症之一。CTD-PAH其临床表现隐匿,早期诊断困难,治疗效果不佳,是CTD患者死亡的重要因素之一[233]。几乎所有的CTD均可出现PAH,常见者包括SLE、SSc、MCTD以及SS等。引起PAH的CTD谱可能存在种族差异,欧美以SSc为主,中国以SLE为主。对于所有PAH患者,均应常规进行CTD筛查。
评估CTD-PAH首先需要确定原发病是否活动,脏器受累是否存在可逆性,借以推断免疫抑制治疗逆转肺血管病变的可能性,从而指导免疫抑制治疗方案的选择。CTD病情活动性评估和危险分层根据不同CTD病种而各有不同,目前主要依据公认的CTD整体活动性评估体系和针对主要受累器官的评分方法,如SSc的皮肤改良Rodnan评分、SLE疾病活动指数(SLEDAI)和不列颠群岛狼疮评估组(BILAG)评分和原发性SS的欧洲抗风湿病联盟疾病活动度指数(ESSDAI)。PAH的病情评估和危险分层详见本指南第五部分“临床严重性评估”。
【推荐意见】
1.推荐疑诊CTD-PAH患者应在风湿免疫科医师的参与下尽早筛查和确诊(1B)。
2.推荐对CTD-PAH的患者进行双重评估:包括CTD病情活动性、PAH严重程度(1C)。
【推荐意见说明】
CTD-PAH的诊断与治疗相对复杂,约半数的患者就诊于医院时,其生活质量已受到明显影响,心功能处于WHO功能分级Ⅲ~Ⅳ级,疾病多数已进入进展期甚至终末期,因此,CTD-PAH的诊断与治疗方案需要风湿免疫科医师的早期参与。
特别需要指出的是虽然无论SLE还是SS、SSc的疾病活动性评分均未将PH纳入评估,但不能片面地将PAH完全隔离出来单独评估,应将PAH作为CTD系统受累的一部分进行全面的综合评估,在临床实践中进行个体化治疗。整体评估疾病活动评分(physician global assessment,PGA)常是风湿科医师最终的评判,通常PGA<1分表示SLE病情处于临床相对缓解状态,而PGA评分增加0.3分提示病情有活动表现。
2.CTD-PAH的治疗:由于自身免疫和炎症反应导致肺血管损伤在CTD-PAH的发生、发展中起到重要作用,尤其在病程早期和病情活动性高的患者中更是这样。因此,除了针对PAH的一般性治疗和靶向药物治疗外,大剂量糖皮质激素联合免疫抑制剂可迅速缓解并稳定基础CTD病情,可有效地改善甚至“治愈”PAH[234, 235]。
CTD-PAH基础病的治疗十分重要,对改善和稳定PAH的病情至关重要。应根据CTD病情是否活动及PAH是否达标来决定相应的治疗方案:糖皮质激素联合免疫抑制剂,包括环磷酰胺、霉酚酸酯、硫唑嘌呤、甲氨蝶呤或羟氯喹等的方案需要个体化,可由风湿科医师确定相应的诱导缓解、巩固缓解和维持缓解的序贯治疗。针对PAH的治疗亦十分重要,应根据PAH的危险分层积极制定最佳的治疗方案。
PAH的治疗原则和策略见指南第五部分相关内容。CTD-PAH治疗的靶目标即最大程度地改善患者预后,提高患者生活质量。短期目标就是延缓临床恶化的时间。我国在CTD-PAH治疗领域率先提出“双重达标治疗”的治疗理念,强调CTD免疫抑制治疗与PAH相关药物治疗并重,对于部分CTD患者,特别是处于疾病早期且病情活动的SLE患者,经过积极的免疫抑制治疗甚至可以实现PAH“治愈”的目标[234]。
CTD治疗目标总体以PGA<1分表示CTD处于临床缓解状态。以三种常见CTD的治疗目标为例:SLE达到低疾病活动度状态,即SLE活动度评分(SLEDAI-2K)≤4分,仅服用抗疟疾药物,可合并联用低剂量糖皮质激素;pSS需达到低疾病活动度状态,即ESSDAI<4分;SSc短期内无进展性皮肤或肺部纤维化病变或血管病变(指端溃疡、PH)可作为治疗目标。
【推荐意见】
推荐CTD-PAH的患者进行“双重达标”治疗:包括CTD相关治疗、PAH治疗(1C)。
【推荐意见说明】
在针对SSc相关PAH的临床研究中发现,mPAP即使轻微升高(21~24 mmHg),也有运动受限的症状、预后不良,并且部分患者会发展为PH[236, 237],针对这部分患者应加强监测随访[49]。
PoPH的临床表现无特异性,与其他类型PAH相似,同时具有门脉高压及原发肝脏疾病的临床表现,最常见的症状是活动后呼吸困难,疾病晚期会出现右心衰竭的相关表现。PoPH的诊断性评估流程与其他类型PAH相同。目前,经胸多普勒超声心动图常作为PoPH的筛查手段。需要完善RHC等相关检查以进一步评估疾病严重程度、监测血流动力学变化和发现其他潜在病因,如肺部疾病、左心疾病或慢性血栓栓塞性疾病。需要注意,同时存在门脉高压和PH并不代表一定是PoPH[2]。
PoPH的治疗策略与其他类型PAH相同,但同时应考虑肝脏疾病的严重程度和药物对肝脏的影响。2016年国际肝脏移植学会发表的最新共识[19]指出:在常规予以靶向药物治疗前,若mPAP<35 mmHg,不增加肝移植围手术期病死率;若mPAP≥35 mmHg,应对PoPH患者进行靶向药物治疗;应将PoPH且mPAP在45~50 mmHg或以上作为肝移植的绝对禁忌证。利奥西呱易在肝内蓄积,不建议用于严重肝损伤(Child-Paugh C级)的患者[238, 239];另外,研究发现马昔腾坦虽然不能改善患者的6MWD、WHO功能分级,但显著降低了PoPH患者的肺血管阻力,且无明显肝毒性等不良反应[240],对于PoPH患者可考虑使用并密切观察、随访。β受体阻滞剂和CCBs可能会造成PoPH患者病情进一步恶化而不推荐使用。PoPH患者通常出血风险较高,因此不建议使用抗凝药物。
【推荐意见】
1.推荐对所有门脉高压及拟行肝移植手术的患者行超声心动图检查以筛查PAH(1B)。
2.推荐PoPH患者采用与其他PAH患者相同的治疗策略(1C),同时应考虑肝脏疾病的严重程度(2C)。不推荐PoPH患者使用抗凝治疗(1C)。建议PoPH患者尽早评估肝移植可能性(2C)。
【推荐意见说明】
目前没有足够的循证医学证据制订PoPH患者的治疗共识。PoPH治疗的总体原则需要以原发肝脏疾病治疗为基础,并积极进行多学科讨论评估肝移植治疗的可能性,少数专科中心可慎重选择患者行肝肺或肝心肺移植[241]。
HIV-PAH的临床表现与IPAH相似,以HIV感染或其他并发症为主而PH的临床表现无特异性。因此,确诊时多为疾病晚期(处于WHO功能 Ⅲ或Ⅳ级)。由于发病率较低,不推荐无症状且无相关危险因素的HIV感染者进行PH筛查,但若存在相关危险因素或出现不明原因的呼吸困难,要进行PH相关检查[2, 100]。
目前HIV-PAH的治疗应遵循PAH的治疗原则,同时给予高效抗逆转录病毒治疗(highly active anti-retroviral therapy, HAART)疗法。研究表明,接受HAART疗法、PAH特异性治疗以及HAART和PAH综合治疗的患者较对照组生存率更高。使用PDE5抑制剂时应注意药物相互作用,因为影响其代谢的酶P450 CYP 3A4和CYP 2C9会被包含利托那韦在内的蛋白酶抑制剂所抑制。HIV-PAH对急性血管扩张剂几乎无反应,因此不建议使用CCBs[242]。由于增加出血风险和药物相互作用,不建议对HIV-PAH患者使用抗凝剂。HIV感染是肺移植手术的禁忌。
PVOD和PCH这两个疾病与IPAH临床上难以区别。
1.PVOD/PCH的诊断:PVOD/PCH和IPAH临床表现极为相似,以往由于诊断意识不高,缺乏确诊手段,绝大多数PVOD/PCH被误诊为IPAH。目前诊断PVOD/PCH需要结合临床表现、影像学特点、肺功能指标、血流动力学指标以及基因检测,必要时需要组织病理学诊断。
(1)影像学表现:X线胸片对PVOD/PCH诊断价值有限,病情严重时可表现为肺水肿。胸部HRCT是诊断PVOD/PCH的主要影像学手段。PVOD/PCH主要影像特征包括:纵隔淋巴结肿大、小叶中心磨玻璃阴影和小叶间隔增厚[243, 244]。一项经组织学证实的研究显示,75%的PVOD/PCH患者至少有上述两种HRCT异常,但缺乏或仅具备一项上述 HRCT征象并不能完全排除PVOD/PCH[244]。
(2)肺功能检查、血气分析:PVOD/PCH尽管在HRCT上存在肺实质异常,但肺容积和肺活量通常保持不变,而DLCO多严重下降,常低于50%预测值。血气分析常显示低氧血症[243, 244]。
(3)RHC:PVOD/PCH的血流动力学特点与其他类型的毛细血管前PH并无不同,虽然PVOD的主要病变位于毛细血管后小静脉,但PAWP通常是正常的。对于PVOD/PCH患者,行急性血管反应试验有发生肺水肿的风险,因此不建议行急性血管反应试验。
主要支持PVOD/PCH诊断的特征有严重的弥散功能障碍、静息时低氧血症、2项及以上HRCT异常特征、支气管肺泡灌洗液可见含铁血黄素细胞。最后,基因检测发现 EIF2AK4双等位基因突变可确诊遗传性PVOD/PCH。
【推荐意见】
推荐疑诊PVOD/PCH患者进行基因检测,如存在EIF2AK4双等位基因突变即可确诊(1B)。
【推荐意见说明】
尽管组织病理检查是PVOD/PCH诊断的金标准,但由于患者常一般情况差,很难耐受肺活检。因此对于疑诊患者,可根据相应的临床及无创性检查特征,做出临床诊断。
2.PVOD/PCH的治疗:(1)一般治疗及支持治疗:对于合并低氧血症的患者可给予氧疗,因PVOD/PCH患者常有潜在出血风险,不建议常规抗凝治疗。其他支持性治疗可参考其他PAH的治疗。
(2)靶向药物治疗:有关靶向药物治疗PVOD/PCH的研究数据很少且相互矛盾。部分PVOD/PCH患者可能通过靶向药物治疗获得短期血流动力学和功能改善,或至少病情稳定,但均缺乏长期疗效。因此,PVOD/PCH患者应用靶向药物必须非常谨慎,且应在有经验的中心严密监护下使用。任何一种靶向药物均有可能引起致命的肺水肿[243]。
(3)免疫治疗:继发于结缔组织病(如SLE、MCTD)或结节病的PVOD/PCH可应用激素及免疫抑制剂治疗。不建议特发性、遗传性及系统性硬化症继发的 PVOD/PCH 患者应用。
(4)肺移植:肺移植或心肺联合移植仍然是PVOD/PCH 患者长期生存的治疗手段,因PVOD/PCH进展迅速,且大多数患者处于晚期疾病,所以,对于符合条件的患者确诊后应尽早转诊进行移植评估[245]。
【推荐意见】
1.推荐PVOD/PCH患者应尽早评估肺移植可能性(1C)。
2.应该警惕PVOD/PCH患者应用靶向药物发生肺水肿,建议将PVOD/PCH患者转诊至PH中心接受进一步评估、治疗及管理(2C)。
【推荐意见说明】
PVOD/PCH患者可能通过靶向药物治疗获得短期改善,但均缺乏长期疗效。因此,肺移植或心肺联合移植仍然是PVOD/PCH患者长期生存的治疗手段,对于符合条件的患者确诊后应尽早进行移植评估。
左心疾病是导致PH的常见原因。左心收缩、舒张功能障碍和/或左心瓣膜疾病是最常见的引起肺动脉压力升高的左心疾病,其病理生理特征为左室充盈压升高,继发左房重塑,肺静脉回流受阻,进一步导致肺静脉压力升高,随着病程进展和压力传导,肺动脉发生血管内皮功能障碍并出现反应性血管收缩、神经内分泌与炎性细胞激活、NO减少、内皮素分泌增加及BNP舒张血管作用降低等病理生理改变促使肺血管重塑,导致PH的发生,进一步限制右室将血液转移到肺动脉的能力,造成右心超负荷及右心功能衰竭,同时通过心室相互依赖性也会再次损害左室的充盈。左心疾病合并PH时症状更为严重,运动能力下降明显,预后更差[69, 246]。
左心疾病所致PH血流动力学诊断标准为静息时mPAP≥25 mmHg,且PAWP>15 mmHg。其区别于其他类型PH的最主要血流动力学特点为PAWP升高,属于毛细血管后PH,再根据PVR将其进一步分为单纯毛细血管后PH和混合性毛细血管后PH [2, 247]。单纯毛细血管后PH又称为被动性PH,占左心疾病所致PH的55%,表现为mPAP和PAWP升高,PVR≤3 WU,提示PH是由肺动脉下游左心压力升高被动传导所致,肺血管结构和功能基本正常。混合性毛细血管后PH又称为反应性PH,表现为mPAP和PAWP升高,PVR>3 WU,提示除外左心压力的传导,肺血管结构和功能本身也已经发生改变。
左心疾病所致PH区别于其他类型PH的最大临床特点为存在左心疾病的临床证据。这类PH患者通常伴有许多左心疾病的征象,如年龄>65岁、肥胖、高血压、冠心病、糖尿病、房颤、左束支阻滞、心血管介入和左心扩大等,根据左心疾病的征象可以预判存在左心疾病的可能性[247]。
劳力性呼吸困难是所有类型PH的共同表现,但左心疾病所致PH较特异的症状表现为端坐呼吸和夜间阵发性呼吸困难。通常结合临床表现、心电图、X线胸片、生物标志物和超声心动图的检查结果可以初步疑诊左心疾病所致PH。
左心疾病所致PH需要进行RHC检查,PAWP或左室舒张末压>15 mmHg常表明存在左心功能不全[70]。当存在左心疾病证据,但静息状态下PAWP≤15 mmHg(特别是应用利尿剂或HFpEF患者)时,常需要在RHC过程中进行(运动或液体)负荷试验可能有助于发现潜在的左心疾病所致PH,但负荷试验的临床意义仍未完全明确[248]。当很难区分是左心疾病所致PH还是第一大类PAH合并左心疾病时,建议将患者转诊至PH中心进一步诊治。
左心疾病所致PH以治疗原发左心疾病为主,包括控制心血管危险因素、药物治疗(包括利尿剂、血管紧张素转化酶抑制剂、β受体阻滞剂等)、非药物治疗(瓣膜置换、冠状动脉再灌注治疗、心室再同步化治疗、左心辅助装置、心脏移植等)以及治疗合并症(COPD、睡眠呼吸暂停综合征、肺栓塞等)。
至今尚没有大样本的随机对照临床试验证实靶向药物可以使左心疾病所致PH患者获益[247, 249]。鉴于此,目前仍不推荐此类患者常规使用靶向药物[2]。今后的临床试验应该建立严格的纳入标准,区分单纯毛细血管后性PH还是混合毛细血管后性PH,筛选出可能对靶向药物敏感的左心疾病所致PH患者。
【推荐意见】
1.推荐左心疾病所致PH患者优化基础疾病的治疗(1B)。
2.经过优化左心疾病治疗后仍存在肺动脉压力明显升高,建议转诊至PH中心进一步诊断和个体化治疗(2C)。
3.不推荐左心疾病所致PH患者常规应用靶向药物(1C)。
【推荐意见说明】
左心疾病所致PH属于毛细血管后PH,经过利尿等药物治疗后仍存在PH者应该进行RHC评估血流动力学,当患者存在mPAP≥25 mmHg, PAWP>15 mmHg,且伴有跨肺压(transpulmonary pressure gradient,TPG)>12 mmHg或肺动脉舒张压差(diastolic pressure gradient,DPG)≥7 mmHg或PVR>3 WU时,建议转诊到有经验的PH中心进一步诊断评估,制定个体化治疗策略。
肺部疾病和(或)低氧所致PH的症状不典型,常与肺部疾病本身所致的胸闷、呼吸困难症状重叠而被掩盖,随着疾病的进展,在肺部疾病体征基础上,可出现PH和右心衰竭的征象。对于临床疑诊PH的肺部疾病患者,需进一步针对PH进行检查。超声心动图是筛查肺部疾病和(或)低氧所致PH的最佳无创检查方法。但由于受到肺气肿的影响,只有38%的COPD患者能够通过检测TRV以估测肺动脉收缩压,对于操作人员的技术要求较高。
慢性肺部或低氧性疾病一般导致轻中度肺动脉压力升高,一旦出现肺动脉压力明显升高(mPAP≥35 mmHg或mPAP≥25 mmHg但伴有低心脏指数<2.0 L/min·m-2)时,需要排查是否合并其他疾病,如左心疾病、CTEPH和PAH等,必要时建议转诊至PH中心进一步诊治。
肺部疾病和(或)低氧所致PH主要针对原发病治疗,推荐长程氧疗,不推荐常规给予靶向药物治疗。
【推荐意见】
1.经过优化肺部疾病治疗后仍存在肺动脉压力明显升高,推荐转诊至PH中心(1C)。
2.对存在慢性低氧的肺部疾病所致PH患者,推荐长期氧疗(1C)。
3.不推荐肺部疾病和(或)低氧所致PH患者常规使用靶向药物(1C)。
【推荐意见说明】
慢性肺部或低氧性疾病合并肺动脉压力升高,需要排查是否合并其他疾病,如左心疾病、CTEPH和PAH等。治疗过程中强调原发病治疗,尤其是长程氧疗的重要性。
CTEPH是以肺动脉血栓机化、肺血管重塑致血管狭窄或闭塞,肺动脉压力进行性升高,最终导致右心功能衰竭为特征的一类疾病。CTEPH属于PH的第四大类,也是可能治愈的一类PH。
CTEPH患者致残率、致死率较高,且生活质量较差,若能早期诊断、及时治疗,将显著改善患者生存状况。因此,理解急性PTE后CTEPH的发生发展过程、识别相关危险因素有助于早期发现CTEPH并进行干预。目前多项研究探讨了PTE后CTEPH发病的危险因素,然而不同研究间筛选出的危险因素不尽相同。年龄、无诱因肺栓塞、复发性肺栓塞、灌注缺损面积较大、近端肺栓塞、初始肺动脉收缩压升高、合并下肢静脉曲张病史等均被证实为CTEPH发病的危险因素[81, 250, 251]。
【推荐意见】
对于急性PTE经规范抗凝3个月后仍有呼吸困难、运动耐量受限的患者建议进行CTEPH筛查(2C)。
【推荐意见说明】
急性PTE规范抗凝治疗后3~6个月应进行常规临床评估:对于急性PTE后持续存在呼吸困难、V/Q显像提示存在灌注缺损且通气灌注不匹配的患者,应对其进行CTEPH诊断评估。目前不建议对急性PTE后无症状且无相关危险因素的患者行CTEPH的常规筛查。
CTEPH最常见的症状是活动后呼吸困难,呈进行性加重,运动耐量下降,其他症状包括咯血、晕厥等。随着病情进展,可出现PH和右心衰竭征象,如口唇发绀、颈静脉怒张、P2亢进、下肢水肿,甚至出现胸腔和腹腔积液等。CTEPH的诊断标准为:经过3个月以上规范抗凝治疗后,影像学证实肺动脉存在慢性血栓,静息状态下RHC测得mPAP≥25 mmHg,且除外其他病变,如血管炎、肺动脉肉瘤、纤维素性纵隔炎等。对于无PH,而存在慢性血栓栓塞的患者,称之为慢性血栓栓塞性疾病(chronic thromboembolic disease, CTED)。
对于临床疑诊或超声心动图检查提示PH的患者,可经进一步检查明确CTEPH的诊断,主要包括肺V/Q显像、CTPA、MRPA、RHC和肺动脉造影等。
1.肺V/Q显像:V/Q显像对CTEPH诊断的敏感性>97%,目前被公认为CTEPH的首选筛查方法,如V/Q显像阴性,可基本排除CTEPH[108, 252, 253]。CTEPH患者V/Q显像的典型表现为多个肺段分布的与通气显像不匹配的放射性灌注缺损。其他原因导致的肺动脉阻塞时也可出现灌注缺损,如肺动脉肉瘤、纤维素性纵隔炎等,需要结合临床及其他影像学资料进行鉴别。
2.CTPA:直接征象包括机化的栓子部分或完全阻塞肺动脉分支,表现为肺动脉内偏心性附壁充盈缺损、肺动脉闭塞、血管腔内线状影或网状纤维化等。间接征象包括PH、右心增大与肥厚、体循环侧支供血增加、肺通气与灌注不匹配导致的马赛克征象以及肺梗死灶等。CTPA对CTEPH的栓塞病变部位、阻塞程度以及右心结构功能评估等有较高价值,同时对于鉴别其他类型肺血管病变具有重要价值,有经验的中心也可通过CTPA评估手术治疗的可行性。由于CTPA检出段以下为主的栓塞性病变敏感性较差,阴性不能完全排除CTEPH诊断[254]。
3.RHC和肺动脉造影检查:通过肺动脉造影检查,可确定慢性血栓栓塞的存在与否,并明确栓塞部位及程度。肺动脉造影可以显示血栓机化和再通,包括肺动脉狭窄或分支闭塞、血管壁不规则、管腔内网状充盈缺损、肺动脉近端扩张与造影剂滞留并远端狭窄等。RHC检测血流动力学指标,用于评估病情和指导治疗。
总体而言,肺V/Q显像通常作为CTEPH诊断的首选筛查手段,肺动脉造影和RHC是CTEPH影像学诊断和手术评估的“金标准”,尽管CTPA检出段以下病变敏感性差,但对于判断近端栓塞的病变部位、程度、手术评估以及鉴别诊断均有重要价值。
【推荐意见】
推荐CTPA作为CTEPH的初筛检查方法(1C);推荐V/Q显像作为CTEPH的除外检查方法(1C);推荐RHC及肺动脉造影作为CTEPH术前评估检查方法(1C)。
【推荐意见说明】
V/Q显像及CTPA均为CTEPH常用的筛查方法。V/Q显像的敏感性显著优于CTPA;V/Q显像阴性可有效排除CTEPH,其敏感性和特异性分别为90%~100%、94%~100%[108, 252]。本指南推荐V/Q显像作为CTEPH除外诊断的首选检查。
CTEPH的治疗包括基础治疗、手术治疗、药物治疗和介入治疗;基础治疗主要包括长期抗凝治疗、家庭氧疗、改善心功能和康复治疗等。
PEA是治疗CTEPH最有效的方法,手术评估需要在有经验的中心进行,部分CTEPH患者可通过手术完全治愈[255]。手术在深低温停循环技术下进行,手术适应证包括:术前WHO功能分级Ⅱ~Ⅳ级,外科手术可及的肺动脉主干、叶或段肺动脉的血栓。高龄、PVR高和右心功能不全影响手术的预后。不能行PEA手术或PEA术后持续性或再发性PH的患者预后较差。PEA手术复杂,围手术期需要呼吸与危重症、心血管、麻醉、体外循环、影像等多学科团队密切合作。
部分CTEPH患者,可行球囊肺动脉成形术(balloon pulmonary angioplasty,BPA)治疗,BPA能明显改善患者症状和血流动力学指标。BPA应在专业的诊疗中心进行。BPA适应证为存在远端慢性血栓栓塞但不宜行PEA术的患者,或者PEA术后存在残余PH或复发性PH的患者。BPA的主要并发症为肺血管损伤和再灌注肺水肿[253, 256]。
虽然PEA是大多数CTEPH患者的治疗选择,但仍有约40%的患者由于血栓位置难以触及而不适合行PEA[257]。sGC激动剂(利奥西呱)等靶向药物可改善CTEPH患者的活动耐力或血流动力学,可用于不能行PEA手术、PEA术后持续或再发的CTEPH患者[258, 259]。利奥西呱是目前唯一获批CTEPH适应证的靶向治疗药物。在不能手术或术后复发/持续肺动脉高压的CTEPH患者中,利奥西呱治疗16周后显著改善了患者的6MWD、WHO功能分级以及血流动力学,在不能手术或术后复发/持续肺动脉高压的CTEPH患者中,利奥西呱治疗长期耐受性和安全性良好,运动和心功能改善持续获益,估测的1年生存率和无临床恶化生存率分别为97%和88%,估测的2年生存率和无临床恶化生存率分别为93%和82%。其他靶向药物在CTEPH的治疗中也有不同程度的获益。CTEPH治疗流程见图4[80]。
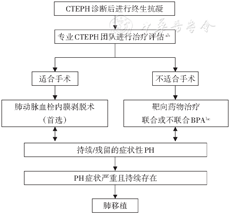
注:PH:肺动脉高压;CTEPH:慢性血栓栓塞性肺动脉高压;BPA:球囊肺动脉成形术;a多学科团队:包括PEA手术专家,PH专家,BPA手术专家和放射科专家;b没有给予靶向药物治疗时也可以考虑BPA;c治疗方案的选择需要取决于专业知识程度
【推荐意见】
1.CTEPH患者若无抗凝禁忌,推荐终生抗凝治疗(1B)。推荐进行手术评估,如能手术,首选PEA(1C)。
2.不能行PEA手术或术后存在持续性或再发性PH患者,推荐应用靶向药物治疗,首选sGC激动剂(利奥西呱)(1B);如具备专业技术条件,建议BPA(2C)。
3.无论是行PEA、BPA,推荐转诊到有条件的中心进行系统评估,评估手段包括:CTPA、V/Q显像、RHC和肺动脉造影等(1C)。
【推荐意见说明】
抗凝治疗可预防静脉血栓栓塞症复发及肺动脉原位血栓形成,防止栓塞病变的进一步加重。因此对于CTEPH患者推荐终生抗凝,抗凝药物通常选择华法林。由于目前缺少直接口服抗凝药物(direct oral anticoagulants,DOACs)在CTEPH的研究证据,故DOACs在CTEPH抗凝治疗中的效果有待进一步评价。
PEA手术是治疗CTEPH最有效的方法,部分CTEPH患者可通过手术完全治愈。该手术技术复杂,需要多学科团队的协作。手术评估需要在有经验的中心进行,对于一个中心评估认为不适合手术治疗的CTEPH患者,推荐到另一家更有手术经验的中心进行第2次评估。
所有确诊的CTEPH患者均应进行PEA手术评估,对于不适合行PEA治疗的患者,可考虑给予BPA或靶向药物治疗。但对于可以行手术治疗的患者,建议不要因为药物治疗而延误手术治疗时机。
第5大类PH为机制未明和(或)多因素共同作用所致,涉及到多个系统的疾病,包括血液性疾病、系统性疾病、代谢性疾病及其他罕见疾病(表15),这些疾病引起PH的机制复杂。在PH病因筛查过程中不要漏诊这些疾病,特别是多系统受累的PH患者;而对于血液性疾病、系统性疾病和代谢性疾病患者应该警惕PH的发生和发展,尽早发现,尽早治疗。这一类PH以治疗原发疾病为主,建议转诊到相应的专科接受治疗,目前不推荐靶向药物用于这一类患者。

末明和(或)多种因素导致的肺动脉高压(PH)
末明和(或)多种因素导致的肺动脉高压(PH)
| 分类 | 亚类 |
|---|---|
| 血液系统疾病 | 慢性溶血性贫血 |
| 骨髓增殖性疾病 | |
| 系统性和代谢性疾病 | 肺朗格汉斯细胞组织细胞增生症 |
| 戈谢病 | |
| 糖原储积症 | |
| 神经纤维瘤病 | |
| 结节病 | |
| 复杂先天性心脏病 | 节段性肺动脉高压 |
| 起源于动脉导管的单侧肺动脉 | |
| 肺动脉缺如 | |
| 肺动脉闭锁伴室间隔缺损和大的主肺侧支动脉 | |
| 肺动脉起源于主动脉 | |
| 其他 | |
| 单心室发育 | |
| 未手术 | |
| 已手术 | |
| 弯刀综合征 | |
| 其他 | 伴或不伴血液透析的慢性肾功能衰竭 |
| 纤维性纵隔炎 |
儿童PH可出现在从新生儿到成人的任何一个年龄段,但不同年龄段的病因与疾病谱不尽相同。鉴于儿童的独特性,本指南将其单独列出进行阐述。儿童PH在病因及病理生理机制等方面与成人有很多相似之处,但儿童也有其自身特点。儿童PH患者的肺血管结构、功能、临床病程、右心室适应性改变以及靶向治疗反应性等方面均与成人患者存在差异,更易受到遗传性与发育性因素的影响。早期识别PH的易感因素,并采取干预措施,部分患儿的PH病变可得到有效逆转。目前,大部分关于儿童PH的临床实践缺乏足够循证医学依据,儿童PH的诊断和治疗策略往往参考成人[260, 261]。
儿童IPAH/HPAH、CHD-PAH和肺发育障碍相关PH比例高于成人,成人PH临床分类是否适合儿童患者仍有争议,但目前国内外儿童PH专家共识仍然采用成人PH的临床分类。
婴幼儿PH患者症状主要表现为呼吸急促、食欲差、发育迟缓和哭闹后紫绀等,年龄稍大的儿童患者临床表现与成人相似。儿童PH的病因复杂,其诊断流程与成人PH相似,但重点在于筛查CHD-PAH、遗传性疾病、肺部或左心发育异常疾病以及代谢性疾病等,对于疑难儿童PH应该转诊到专业的儿童PH中心诊治[261]。
超声心动图操作简便无创、可重复,是儿童PH筛查和随访最重要的无创性检查手段。CTA和MRI可对心脏大血管结构、肺部疾病和心脏功能进行评估。RHC仍然是儿童PH诊断的金标准,相关血流动力学参数可用于评估疾病严重程度并为风险分层提供依据[262, 263]。常规化验、自身抗体和血、尿代谢指标等检查有助于发现PH的潜在病因。BNP和NT-proBNP是评价PH患者右心功能和病情严重程度、指导临床决策的重要指标。染色体异常与遗传因素在儿童PAH发病中起着重要作用,BMPR2、ACVRL1、CAV1和KCNK3等基因突变与IPAH/HPAH相关,EIF2AK4基因突变与PVOD有关,进行相关基因检测可以有助于明确诊断、评估预后、指导生育。
【推荐意见】
1.推荐疑诊PH的患儿在专业儿童PH中心就诊,尽量依据诊断流程进行全面评估(1B)。
2.推荐诊断PAH及开始靶向治疗前行RHC,必要时可行急性血管反应试验(1B)。
3.推荐大龄PH儿童行心肺运动试验和6MWT以评估运动耐量、治疗效果及预后(1B)。
【推荐意见说明】
RHC在明确诊断、判断疾病严重程度、指导治疗方面起决定性作用。考虑到儿童患者的特征以及RHC检查存在的风险,建议在经验丰富的小儿心血管中心实施RHC检查。急性血管反应试验可用于评估IPAH/HPAH儿童患者接受CCBs治疗指征,以及CHD-PAH患者能否接受心脏畸形矫治手术。目前急性血管反应试验药物仍首选吸入NO,还可选用吸氧以及雾化吸入伊洛前列素,伊洛前列素剂量为0.3~0.5 μg/kg,持续吸入10 min[264]。如果采用Fick法测定肺血流量,氧气吸入可能会影响PVR的准确性。IPAH/HPAH儿童的急性血管反应试验阳性标准仍参照成人标准,儿童患者阳性率为8%~15%。需要注意的是,同成人一样,口服CCBs的儿童也需要密切随访,定期重复RHC以保证急性血管反应试验持续阳性。存在CHD分流的患者,由于缺乏血流动力学及急性血管反应试验预测手术效果及预后的研究,采用何种阳性标准存在争议。通常认为,PVRI下降幅度超过20%,同时最终的PVRI< 6 WU·m2并且PVRI/SVRI<0.3可能是较好的CHD矫治手术指征。
1.治疗目标:2018年第六届WSPH专家建议采用儿童版PAH危险分层,治疗靶目标是达到低危状态,治疗的最终目标应该是提高患儿存活率,促进患儿不受限制的正常活动[260]。
2.治疗策略:不同类型的儿童PH治疗策略不同,建议确诊为PH的患儿转诊到专科医疗机构接受规范治疗。在强心、利尿和吸氧等一般治疗的基础上,在无明确禁忌证的前提下先行RHC和必要的急性血管反应试验。
由于针对儿童PAH治疗的循证医学证据有限,主要参照成人PAH的治疗策略(图3)。靶向药物缺乏儿童专用剂型,儿童PAH需按照千克体重给药。对于年龄>1岁并且急性血管反应试验阳性的PAH患儿,可选用CCBs,在服用CCBs后临床改善并持续阳性者,可以继续应用CCBs;如果出现临床恶化,则需要再次进行评估并调整治疗方案。对于急性血管反应试验阴性以及CCBs治疗效果不好者,则需要根据PAH危险分层制定相应的治疗方案:对于低危患者,可首选ERA(如波生坦)或PDE5抑制剂单药治疗,2011年欧洲药监局批准西地那非应用于1~17岁的儿童PAH,推荐剂量为:年龄<1岁,0.5~1 mg/kg每日3次;体重<20 kg,10 mg,每日3次;体重>20 kg,20 mg,每日3次。2009年欧洲批准波生坦用于≥2岁儿童PAH的治疗,推荐剂量为2 mg/kg,后将年龄放宽至1岁[20]。2017年美国食品药品管理局批准了波生坦用于≥3岁 的IPAH及CHD-PAH患者[265]。2019年我国批准波生坦用于≥3岁PAH患儿,儿童推荐剂量见表16。如果靶向药物效果不好但临床评估后仍属于低危状态,也可试用雾化吸入前列环素类似物或前列环素受体激动剂。单药治疗后临床恶化者需要考虑早期联合靶向药物治疗。高危PAH患儿静脉滴注依前列醇或曲前列尼尔为首选治疗方案,也可考虑皮下注射曲前列尼尔或早期联合靶向药物治疗。在最大限度的药物治疗后病情仍然恶化的患儿,则考虑房间隔造口术、Potts分流术或肺/心肺移植术。
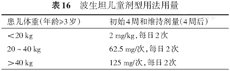
波生坦儿童剂型用法用量
波生坦儿童剂型用法用量
| 患儿体重(年龄≥3岁) | 初始4周和维持剂量(4周后) |
|---|---|
| <20 kg | 2 mg/kg,每日2次 |
| 20~40 kg | 62.5 mg/次,每日2次 |
| >40 kg | 125 mg/次,每日2次 |
【推荐意见】
1.推荐对IPAH/HPAH患儿进行危险度分层,以维持低危作为治疗目标(1B)。
2.推荐儿童PAH应用波生坦儿童剂型(1B);使用伊前列醇、曲前列尼尔可改善PAH患儿血流动力学参数和生活质量,提高其生存率(2B)。
3.建议靶向药物单药治疗效果不佳的儿童PAH患者早期联合治疗(2B)。
4.不推荐儿童PAH接受高剂量口服西地那非治疗(1B)。
【推荐意见说明】
年龄>1岁且急性血管反应试验持续阳性的儿童IPAH/HPAH,可口服CCBs。对于阴性且风险较低的儿童PAH,建议开始口服单个靶向药物治疗,包括ERA(波生坦)或PDE5抑制剂(西地那非、他达拉非)。与成人PAH患者一样,儿童高风险的决定因素包括右心室衰竭的临床证据、症状的进展、WHO功能分级Ⅲ或Ⅳ级、BNP/NT-proBNP水平显著升高、严重右心室功能障碍和心包积液。高风险的PAH患儿建议积极早期靶向药物联合治疗。
近年来,虽然PH认识逐渐加深,但PH患者的诊断与治疗现状仍不容乐观[51, 63]。诊断困难的主要原因与PH症状隐袭、缺乏特异性、病因涉及多学科等有关[266],早期识别与及时转诊至具有综合诊治能力的PH中心,有助于正确诊断和充分治疗。随着越来越多的靶向药物进入我国市场,不少患者没有经过全面病因筛查和功能评估便使用了靶向药物治疗,这导致了PH患者医疗照护模式不统一,对指南的遵循程度以及专业水平也不一致。以PAH为代表的肺血管疾病相对少见,且临床诊治相对复杂,总体而言,患者集中在PH中心诊治效果更好,专业的有经验的PH中心在诊治效果、节约诊疗成本与获得患者团体支持等方面均有突出优势[2]。
PH中心建设至关重要,一方面能够提高诊治效果,同时也能够节约成本。建立PH中心的目的主要在于接收某些需要特定方法诊断和需要使用特定药物治疗的PAH、CTEPH、其他疑难危重症PH患者,以及对可引起PH的所有病因进行系统研究与评估。PH中心的建设对于改善患者的结局和预后具有重要的价值。
建议所有成人PH患者首先进行ECG、X线胸片、超声心动图和肺功能等初步检查,经过心内科或呼吸科医生会诊后,转诊至PH中心[267, 268]。PH是一种进展性疾病,一旦病情恶化,转诊至PH中心,常能接受更好的综合救治措施[267, 268]。
PH中心应具备较强的临床诊治能力,比如欧美国家的PH中心要求年接诊PH病例数不少于200例,年随访PAH或CTEPH患者不少于50例,每月平均新增PAH或CTEPH患者不少于5例,具备RHC检查技术且进行急性血管反应试验例数每年不少于20例,要求有多学科组成的专家团队,配备专科病房与门诊以及相关检查与药物,并且有合作网络、教育培训等,这些经验与要求对于我国PH中心的建设有着很好的借鉴作用,但中国PH患者基数大、医疗资源地域分布差异有别于欧美国家,我国PH中心标准的建立还需要结合我国特点进一步摸索。
PH患者转诊至PH中心的指征包括:PAH、CTEPH及各种疑难危重的PH患者等;当慢性肺部疾病或左心疾病患者出现明显PH不能用基础疾病解释或严重右心衰竭时也应该考虑转诊;当儿童PH出现下列情况应转诊至儿童PH中心:确诊或怀疑IPAH或家族性PH、新生儿持续性PH、术后持续性PH、不能手术的CHD相关PAH、ILD相关PH、多因素导致的PH以及不明原因的PH。
指南制定成员(按单位名称汉语拼音排序)
指南制定组长:中国医学科学院 北京协和医学院 呼吸病学研究院 国家呼吸医学中心(王辰)
顾问:中国医学科学院阜外医院肺血管病诊治中心(程显声),冠心病诊治中心(高润霖);广州医科大学附属第一医院 广州呼吸健康研究院 国家呼吸医学中心 国家呼吸系统疾病临床医学研究中心 呼吸疾病国家重点实验室(钟南山)
外籍顾问:美国加利福尼亚大学圣地亚哥分校医学院(Jason X._J. Yuan);英国帝国理工大学医学院(Lan Zhao)
统稿专家组:
组长:中日友好医院呼吸中心 呼吸与危重症医学科 国家呼吸医学中心 国家呼吸系统疾病临床医学研究中心(翟振国)
主要成员:中国医学科学院阜外医院肺血管病诊治中心(熊长明);兰州大学健康数据科学研究院 循证医学中心(陈耀龙);首都医科大学附属北京朝阳医院呼吸与危重症医学科(杨媛华);中日友好医院呼吸中心呼吸与危重症医学科 国家呼吸系统疾病临床医学研究中心(万钧)
执笔专家组:
组长:北部战区总医院呼吸与危重症医学科(马壮)
主要成员:北部战区总医院先心病内科(王琦光、朱鲜阳);北京协和医院风湿免疫科(李梦涛、王迁、赵久良、曾小峰);北京协和医院呼吸与危重症医学科(施举红);北京医院呼吸与危重症医学科(许小毛);重庆医科大学附属第一医院呼吸与危重症医学科(陈虹);大连医科大学附属第一医院呼吸与危重症医学科(季颖群、张中和);中国医学科学院阜外医院肺血管病诊治中心(罗勤、柳志红、熊长明);广州医科大学附属第一医院 广州呼吸健康研究院 国家呼吸系统疾病临床医学研究中心 国家呼吸医学中心 呼吸疾病国家重点实验室(王健),呼吸与危重症医学科(洪城);华中科技大学同济医学院病理生理学系(胡清华);四川大学华西医院呼吸与危重症医学科(易群);四川省人民医院呼吸与危重症医学科(郭璐);首都医科大学附属北京安贞医院小儿心脏中心 (顾虹);首都医科大学附属北京朝阳医院呼吸与危重症医学科(杨媛华);上海交通大学医学院附属上海儿童医学中心(徐卓明);同济大学附属上海市肺科医院肺循环科(宫素岗、刘锦铭);无锡市人民医院呼吸与危重症医学科(吴波);中日友好医院呼吸中心呼吸与危重症医学科 国家呼吸系统疾病临床医学研究中心(陈文慧、陶新曹、万钧、谢万木、翟振国),放射诊断科(刘敏)
外审专家组:
组长:中南大学湘雅医院呼吸与危重症医学科(胡成平)
主要成员:北部战区总医院呼吸与危重症医学科(刘蕾);蚌埠医学院第一附属医院呼吸与危重症医学科(许启霞);北京大学国际医院呼吸与危重症医学科(刘双);北京大学第三医院呼吸与危重症医学科(张静);北京大学第一医院儿科(杜军保);北京积水潭医院呼吸与危重症医学科(张运剑)北京医院分子诊断中心(肖飞);重庆医科大学附属第一医院心血管内科(黄玮);东南大学附属中大医院呼吸与危重症医学科(章锐锋);复旦大学附属上海华山医院呼吸与危重症医学科(李圣青);复旦大学附属中山医院心血管内科(周达新、管丽华);福建医科大学附属第一医院呼吸与危重症医学科(邓朝胜);广东省东莞市人民医院呼吸与危重症医学科(张平、徐汝洪);广东省人民医院心血管内科(黄奕高、姚桦、张曹进);广州医科大学附属第一医院呼吸与危重症医学科 广州呼吸健康研究院 国家呼吸系统疾病临床医学研究中心 国家呼吸医学中心(刘春丽、张挪富);广州中医药大学第一附属医院呼吸内科(张伟);甘肃省人民医院心血管内科(曹云山);贵州省人民医院呼吸与危重症医学科(刘维佳);河北医科大学第三医院呼吸与危重症医学科(陈刚);河北医科大学第二医院呼吸与危重症医学科(袁雅冬);河北医科大学第一医院呼吸与危重症医学科(张瑛琪);哈尔滨医科大学药学院(朱大岭);河南科技大学第一附属医院呼吸与危重症医学科(毛毅敏);河南省人民医院呼吸与危重症医学科(赵丽敏);湖南中医药大学(戴爱国);华中科技大学同济医学院附属同济医院呼吸与危重症医学科(刘先胜、甄国华);吉林大学第二医院呼吸与危重症医学科(董春玲、徐伟);江汉大学附属医院呼吸与危重症医学科(李承红);江苏省人民医院呼吸与危重症医学科(解卫平);空军军医大学唐都医院呼吸与危重症医学科(王宁);空军军医大学西京医院呼吸与危重症医学科(张艰);云南大学附属医院呼吸与危重症医学科(邢西迁);内蒙古医科大学第三附属医院内蒙古包钢医院呼吸与危重症医学科(徐喜媛);内蒙古自治区人民医院呼吸与危重症医学科(王佳烈);南通大学附属医院呼吸与危重症医学科(倪松石);青岛大学附属医院呼吸与危重症医学科(程兆忠);首都医科大学附属北京安贞医院 北京市心肺血管疾病研究所(杜杰),呼吸与危重症医学科 (朱光发),急诊危重症中心(聂绍平);山东大学齐鲁医院心血管内科(纪求尚);山东省立医院呼吸与危重症医学科(朱玲);首都医科大学附属北京同仁医院呼吸与危重症医学科(刘广杰);山西省人民医院呼吸与危重症医学科(杜永成、张爱珍);上海交通大学医学院附属仁济医院心血管内科(沈节艳),呼吸科(吴学玲);山西医科大学第一医院呼吸与危重症医学科(胡晓芸、施熠炜);沈阳医学院附属中心医院呼吸与危重症医学科(夏书月);深圳大学生命与海洋科学学院(苟德明);深圳市人民医院呼吸与危重症医学科(傅应云);天津医科大学总医院呼吸与危重症医学科(董丽霞);天津市海河医院呼吸与危重症医学科(吴琦);天津医科大学基础医学院(余鹰);武汉亚洲心脏病医院心血管外科(张刚成);温州医学院附属第一医院呼吸与危重症医学科(王良兴);西安交通大学医学院第一附属医院呼吸与危重症医学科(李满祥),心血管内科(范粉灵);新疆维吾尔自治区人民医院呼吸与危重症医学科(陈颖);新疆维吾尔自治区中医院呼吸与危重症医学科(杨惠琴);新疆医科大学第一附属医院呼吸与危重症医学科(刘晖);新疆医科大学附属肿瘤医院(罗琴);西南医科大学附属医院呼吸与危重症医学科(李玉英);浙江大学医学院附属第一医院高级病房(潘慧云);中国人民解放军总医院第一医学中心呼吸内科(张伟华);中国医科大学附属第一医院呼吸内科(刘璠);中国医科大学附属盛京医院呼吸与危重症医学科(张坡);中国医学科学院阜外医院肺血管诊治中心(何建国),心血管外科(刘盛),实验诊断中心(周洲);中南大学湘雅二医院呼吸与危重症医学科(吴尚洁),心血管内科(李江);中南大学湘雅医院呼吸与危重症医学科(唐勇军、杨威);浙江大学医学院附属邵逸夫医院呼吸与危重症医学科(应可净);浙江大学医学院基础医学院(杨隽);中日友好医院呼吸中心呼吸与危重症医学科 国家呼吸系统疾病临床医学研究中心 心脏血管外科(刘鹏、甄雅南),心脏内科(孙艺红、任景怡);郑州大学第一附属医院呼吸与危重症医学科(程哲、靳建军),急诊医学部(孙同文);
证据评价组:
组长:兰州大学健康数据科学研究院 循证医学中心(陈耀龙)
主要成员:北京医院呼吸与危重症医学科 (王静);兰州大学健康数据科学研究院 循证医学中心(罗旭飞);首都医科大学附属北京安贞医院呼吸与危重症医学科 (张萌);中日友好医院呼吸中心呼吸与危重症医学科 国家呼吸系统疾病临床医学研究中心(陈新旺、陶新曹、高倩、张帅、张云霞、张竹)
秘书组:
组长:中日友好医院呼吸中心呼吸与危重症医学科 国家呼吸系统疾病临床医学研究中心(万钧)
主要成员:北京医院呼吸与危重症医学科 (王静);首都医科大学附属北京安贞医院呼吸与危重症医学科 (张萌);中日友好医院呼吸中心呼吸与危重症医学科 国家呼吸系统疾病临床医学研究中心(谢万木)
所有作者均声明不存在利益冲突

























