
血友病是一种罕见的遗传性出血性疾病,患者临床表现严重程度不一,重型患者致残率高,且治疗费用昂贵,影响患者生存质量。胚胎着床前遗传学检测(PGT)是一种预防遗传病患儿出生的有效手段。为了规范和推广血友病的PGT技术,中国医师协会生殖专业委员会精准辅助生殖研究学组、中国医师协会医学遗传医师分会及血液科专家组成专家委员会,共同讨论和制定了PGT阻断血友病的专家共识,供临床应用参考。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
血友病(hemophilia)是一种X染色体连锁的隐性遗传性出血性疾病,主要分为血友病A、B两型,分别为凝血因子Ⅷ(FⅧ)和Ⅸ(FⅨ)基因突变引起其量的缺乏或质的异常所致。其中血友病A占80%~85%,血友病B占15%~20%。流行病学调查显示,血友病的发病率没有种族和地区差异,男性人群中,血友病A的发病率约2‱,B型约为0.4‱[1, 2, 3, 4]。血友病患者的治疗原则主要遵循按需治疗,在有出血时可及时止血并给予足量替代治疗。血友病可发生颅内出血、血友病性关节病和假性肿瘤等严重并发症,目前针对血友病患者进行长期规律性频繁地诱导免疫耐受治疗(immune tolerance induction,ITI)仍是清除血友病伴抑制物的主要治疗方案,给患者带来极大的痛苦与经济负担,因此积极开展血友病发生的预防工作很有必要[4]。胚胎着床前遗传学检测(preimplantation genetic testing,PGT)是一种有效的预防血友病患儿出生的生育选择。为了规范和推广血友病的PGT技术,由我国血液学、妇产科学和遗传学专家组成专家委员会,结合国内外PGT技术的发展现状,共同讨论和制定本专家共识,供临床应用参考[5]。
血友病A和血友病B临床表现相似,主要表现为自发性出血或轻微损伤后出血不止,以关节出血较为多见,且体表和体内任何部位均有出血风险,可累及皮肤、黏膜、肌肉或脏器。部分重型患者反复出血可致残、致死[1, 2, 3]。根据凝血因子活性水平将血友病分为轻型、中型和重型[1]。
1.筛选试验:血小板计数正常,凝血酶原时间(prothrombin time,PT)正常,凝血酶时间(thrombin time,TT)正常,纤维蛋白原定量正常和激活的部分凝血活酶时间(activated partial thromboplastin time,APTT)不同程度延长[1, 2, 3]。
2.纠正试验:延长的APTT若不能被正常血浆所纠正,可能为获得性血友病或血友病合并凝血因子抑制物[1, 2, 3]。
3.鉴别试验:血友病患者出血时间正常,血管性血友病因子抗原(von Willebrand factor,VWF:Ag)及活性均正常,可以与血管性血友病鉴别。若疑为获得性血友病,在纠正试验的基础上,还应关注抑制物滴度的检测[1, 2, 3]。
4.确诊试验:采用一期法或发色底物法测定凝血因子活性。血友病A患者凝血因子Ⅷ的活性(FⅧ:C)减低或缺乏,VWF:Ag正常,血友病B患者FⅨ:C减低或缺乏[1, 2, 3, 6, 7]。
血友病A和血友病B均为X-连锁隐性遗传的单基因病,致病机制为位于X染色体长臂末端的F8或F9存在基因致病性变异。携带致病性变异的男性为患者,女性通常为携带者,女性患者极为罕见。建议对血友病患者进行基因检测,确定致病基因变异类型后,可为患者本人及同一家族中其他基因突变携带者的产前诊断及胚胎着床前检测提供依据[1, 2, 3, 8]。
重型血友病A患者建议首先进行F8内含子22倒位和内含子1倒位的检测,检测阳性可明确诊断。轻、中型血友病患者以及倒位检测结果为阴性的重型血友病患者,需要进行F8外显子核苷酸测序。倒位检测结果阴性且核苷酸测序未找到致病性突变的患者,需要进行拷贝数变异的检测以明确诊断[1, 2, 3]。血友病B的患者需行F9外显子测序或拷贝数变异检测。
血友病主要分为A、B两个类型,致病基因分别为F8、F9,分别定位于Xq28和Xq27.1(表1)。从世界各地几千例血友病患者中检测出的大量突变来看,血友病的突变分子基础极为多样。血友病A的主要变异类型有倒位/重组、点突变、插入/缺失[9, 10, 11, 12, 13, 14, 15, 16, 17],血友病B的主要变异类型有点突变、缺失/插入[18, 19, 20, 21, 22]。
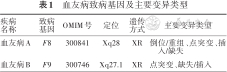
血友病致病基因及主要变异类型
血友病致病基因及主要变异类型
疾病 名称 | 致病基因 | OMIM号 | 定位 | 遗传 方式 | 主要变异类型 |
|---|---|---|---|---|---|
| 血友病A | F8 | 300841 | Xq28 | XR | 倒位/重组、点突变、插入/缺失 |
| 血友病B | F9 | 300746 | Xq27.1 | XR | 点突变、缺失/插入 |
应根据需求选取适当的检测方法。血友病的基因检测方法主要有:
1.长距离聚合酶链式反应(long-distance polymerase chain reaction,LD-PCR)和双管多重PCR:仅需一步PCR可快速、便捷地检测出F8内含子22倒位或内含子1倒位,是血友病A患者与携带者基因变异的首选筛查方法[23, 24, 25, 26]。
2.外显子核苷酸测序:设计特异性引物扩增外显子区域,行Sanger测序,主要针对点突变以及小片段插入/缺失进行检测。
3.高通量测序技术(next-generation sequencing,NGS):能够快速、高效地检测出已知或未知的变异。高通量测序检出的变异位点需Sanger测序验证[27]。
4.多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA):LD-PCR、PCR和NGS结合Sanger检测阴性时,建议MLPA补充检测,主要用于F8、F9部分外显子缺失/重复的检测[14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28]。
1.具有遗传咨询资质的临床医师对患者进行遗传咨询和生育指导。
2.解读基因检测报告上提示的基因与变异位点致病性,结合患者临床表现,判断基因检测结果的有效性,提供疾病发展特点、治疗方法等信息。如检测结果与患者临床表现不符,给出相应建议[29]。
3.为其家系中致病性变异携带者提供遗传咨询和生育指导。理论上女性携带者的子代中:女性50%为携带者,50%为正常;男性50%为血友病患者,50%为正常。男性血友病患者的子代中:女性均为携带者,男性均为正常。咨询中应告知现阶段可采用的医学干预措施(如产前诊断和PGT)及其利弊。夫妻双方在充分了解相关信息的前提下,自愿选择适宜的遗传阻断方式[29, 30, 31]。
4.血友病男性患者一般具有典型临床表现,女性携带者通常无临床表现,但是由于女性携带者存在正常X染色体失活的可能性[32, 33],在X染色体失活极度偏倚的情况下,极少数女性携带者会出现相关临床表现。
PGT是在体外受精-胚胎移植(invitro fertilization-embryo transfer,IVF-ET)技术的基础上,为有生育血友病患儿高风险的夫妻提供胚胎遗传学检测,选择不受累该疾病的胚胎进行移植。
在血友病的致病基因中F8基因变异最为常见也最为复杂,以下内容仅以血友病A为例进行阐述。
重型血友病A患者反复出血可能致残、致死,且FⅧ制剂替代治疗费用昂贵,需要终生治疗,给家庭和社会带来沉重负担。对于具有生育重型血友病患儿高风险的夫妻,如先证者临床诊断明确,且基因致病性变异明确,则该家系满足PGT指征。同时夫妻双方应通过生育力评估,且无其他PGT禁忌证。
夫妻双方外周血检测F8未见异常,如仅有一次血友病患儿生育史,首先推荐产前基因诊断。如有两次及以上血友病患儿生育或妊娠史,不排除生殖腺嵌合的可能性,符合PGT适应证。
经遗传咨询和生育指导后,夫妻双方充分了解PGT的目的和技术局限性以及检测风险,接受并自愿选择进行PGT。
夫妻双方进行PGT前,需首先接受临床遗传咨询,充分了解血友病A的遗传特点、临床表现、目前已有的治疗方案以及子代遗传风险,知晓现阶段可能的遗传阻断措施(如产前诊断、胚胎着床前诊断等)及其利弊,同时需结合生育风险评估结果,夫妻双方知情后自愿选择,并签署知情同意书。如携带突变的女方为新发突变(de novo),或缺乏必需的家系成员(即家系连锁分析所必需),需告知患者PGT的检测策略与风险。
PGT后需告知患者胚胎的致病变异携带情况和可能的风险。血友病A为X-连锁隐性遗传病,需告知携带致病性变异的男性胚胎,不予移植;携带基因突变位点的女性胚胎,理论上不致病,但其男性后代患病风险为50%,夫妻双方需经过遗传咨询,经慎重考虑,签署知情同意书后方可移植杂合携带突变的女胚。
血友病A的PGT目标基因为F8,其常见的变异类型有倒位、点突变(错义突变、无义突变、剪切突变)和插入/缺失[34]。目前PGT检测采用变异位点直接检测联合家系连锁分析的检测策略,(目前F8的1号和22号内含子倒位在胚胎单细胞水平上检测困难,以家系连锁分析为主要诊断策略),理论上对致病基因的变异类型无特殊限制[35]。
1.原则要求:(1)检测前验证的原则要求:首先针对患者的分子遗传检测报告信息在家系成员中评估,并进行家系连锁分析评估,并在单细胞水平评估检测方案的有效性,确定PGT的可行性,为后续的胚胎诊断做准备[29]。(2)胚胎检测的原则要求:胚胎F8变异的直接检测联合遗传连锁分析,连锁分析可选短串联重复序列(short tandem repeat,STR)或单核苷酸多态性(single nucleotide polymorphisms,SNP),以避免因扩增失败、优势扩增、等位基因脱扣(allele drop-out,ADO)等因素所导致的误诊或诊断不明。对于F8内含子22倒位或大片段插入/缺失等特殊的变异形式,单细胞水平难以直接检测,则主要依赖连锁分析进行胚胎诊断[35]。
2.胚胎活检策略:推荐胚胎发育到囊胚阶段显微操作活检获得外滋养层细胞。
胚胎活检样本可在不进行全基因组扩增的情况下进行基因变异检测[36],但检测失败率相对较高。建议对胚胎活检样本进行单细胞全基因组扩增(single-cell whole-genome amplification,WGA),增加DNA的总量以满足多种检测策略的联合使用。目前常用的全基因组扩增技术主要有多重置换扩增(multiple displacement amplification,MDA)、多次退火环状循环扩增(multiple annealing and looping-based amplification cycles,MALBAC)和简并寡核苷酸引物PCR(degenerate oligonucleotide primed PCR,DOP-PCR)[37, 38]。推荐直接突变位点检测与连锁分析及染色体倍性分析联合的策略。在胚胎单细胞水平难以进行直接位点检测并缺乏家系成员的情况下,可以采用性别选择的策略,但应告知患者风险和利弊。
1.变异位点直接检测:根据血友病患者基因检测报告以及检测前验证的结果,对胚胎活检样本进行变异位点直接检测,检测方法主要有:(1)Sanger测序:主要适用于血友病致病变异为点突变、小片段插入/缺失突变的胚胎检测[36]。通过特异性PCR扩增和测序,可以直接检测胚胎的变异位点,但是该方法的胚胎脱扣率、检测失败率较高,单独使用检测风险较大。(2)限制性核酸内切酶法:采用限制性核酸内切酶对PCR扩增产物进行酶切。如变异位点产生/破坏限制性酶切位点,则产生不同长度的片段。该方法局限性大,且误诊可能性高,单独检测风险大[8]。(3)双突变扩增阻滞系统(amplification refractory mutation system,D-ARMS):该方法主要用于血友病致病基因变异为点突变、小片段插入/缺失突变的胚胎检测。该方法可以快速、直接检测已知变异,但对引物设计的要求高[39]。(4)实时荧光定量PCR(quantitative real-time PCR,qPCR):用于点突变、小片段缺失/插入形式血友病家系的胚胎诊断,qPCR需要专门的仪器,对活检样本质量要求高[40]。
2.连锁分析:F8的某些特殊变异形式,如内含子22倒位等,直接在单细胞水平检测相对困难,间接的连锁分析具有显著的优势。基于STR/SNP的连锁分析处可以区分胚胎致病变异的携带情况、可以判断目标基因附近是否发生染色体重组、检测体系是否存在污染等问题。一般要求在变异位点上下游1 Mb内各选择不少于2个可用的SNP或STR位点用于分析[35, 41, 42, 43]。由于F8位于X染色体长臂末端,可用多态性位点较少,所以应提前告知患者仅依靠连锁分析可能出现误诊的风险。
对于缺乏必需家庭成员用于连锁分析(家系不全)的病例,可以利用配子作为分析依据,前提是配子单细胞可检测致病变异位点。由于内含子22倒位等变异形式,单细胞水平很难检出,携带此类变异的夫妻,如果家系不全,不建议进行PGT。
3.性别选择的策略:血友病A为X-连锁隐性遗传疾病,早期针对该病的PGT方法集中于胚胎性别的选择,通过PCR、荧光原位杂交技术等方法选择女性胚胎移植,从而避免子代发生血友病,但移植的女胚中有50%为携带者[30, 32, 37]。除此之外,该策略导致全部男性胚胎均被丢弃(50%的可能为正常胚胎),降低了PGT患者的成功率,因此不做首选推荐策略。对携带内含子倒位或大片段插入/缺失等特殊的变异形式的夫妻,如家系不全,可采用性别选择策略,但需向患者充分交代此检测策略的缺点。
4.胚胎非整倍性检测:除变异位点直接检测和连锁分析,推荐对胚胎进行非整倍性检测,以提高胚胎着床率,降低胎儿流产率以及避免染色体异常患儿出生。非整倍性检测可以采用微阵列比较基因组杂交技术(array-comparative genomic hybridization,aCGH)、SNP微阵列芯片技术(SNP array)和NGS[38, 44, 45, 46, 47, 48]。
1.胚胎检测结果需要两人独立分析判读,最后由第三人审核。诊断不明确的胚胎不建议移植[38],某些特殊情况下:如受检胚胎存在染色体嵌合、或者在基因上下游或基因内部存在染色体重组现象,连锁分析难以判断基因突变携带与否、或者由于扩增失败或检测失败不能得出准确的检测结果等,如患者没有其他胚胎可以选择,胚胎移植可采用性别选择的策略。
2.医疗机构向患者夫妻解读胚胎检测结果。
3.对于检测结果应告知其由于技术局限性不排除假阴性或假阳性可能。
4.对于携带杂合致病性变异的女性胚胎,移植前应充分告知由该胚胎获得的女孩成长到孕龄期生育后代的患病风险。由于女性携带者有出现正常X染色体失活的可能性,在X染色体失活极度偏倚的情况下,部分女性携带者可以出现相关临床表现。患者移植前应充分知情同意。
5.对于染色体嵌合或检测结果判读有风险的胚胎,经专家会诊且患者充分知情同意后,再决定是否可作为备选移植胚胎。
6.建议进行单胚胎移植。
7.检测报告应包含患者基本信息、基因突变信息与胚胎检测结果,注明是否可以移植,特殊情况下需要患者签署胚胎移植知情同意书。
目前尚无明确的证据表明PGT中包括的胚胎活检技术、囊胚培养技术和冷冻复苏技术会增加新生儿不良结局的风险,但其长期影响尚不确定。评价PGT遗传学检测是否增加子代患病风险,建议对PGT子代进行长期随访[49, 50, 51]。
对胚胎移植后PGT的患者,建议宫内妊娠病例要持续随访孕期情况、产前诊断结果等信息。所有PGT后妊娠的患者都应进行产前诊断,降低胚胎检测的可能风险。
对PGT子代随访指标包括但不限于以下方面:出生日期、单胎与多胎及绒毛膜状态、分娩孕周、分娩方式、出生身长及体重、性别、是否存在出生缺陷和出生后定期进行生长发育评估(包括营养、骨骼、神经和运动等)。
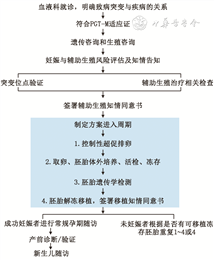
PGT的一般临床流程包括:生殖和遗传咨询、患者入组、体外受精和胚胎活检、遗传学检测、胚胎移植、产前诊断和新生儿随访等方面,具体临床流程见图1。因患者的生育力、基因突变类型等情况不同,诊疗需求存在差异化,因此,在遵循基本临床流程的基础上,对特殊患者需提供个性化诊疗策略。质量管理需从机构人员资质、实验室环境、仪器设备、试剂耗材、实验检测过程、结果判定、报告内容及解读等多个环节进行全程质量控制管理。
执笔者(按姓氏汉语拼音排序):闫丽盈(北京大学第三医院生殖医学科);刘平(北京大学第三医院妇产科)
参与本共识编写、审定的专家(按姓氏汉语拼音排序):曹云霞(安徽医科大学第一附属医院生殖医学中心);陈子江(山东大学 生殖医学研究中心);黄荷凤(复旦大学附属妇产科医院妇科内分泌与生殖医学科);黄锦(北京大学第三医院生殖医学科);景红梅(北京大学第三医院血液科);林戈(中南大学中信湘雅生殖与遗传专科医院);刘平(北京大学第三医院妇产科);卢美松(哈尔滨医科大学附属第一医院妇产科);乔杰(北京大学第三医院妇产科);王学峰(上海瑞金医院血液科);王子莲(中山大学附属第一医院妇产科);邬玲仟(中南大学湘雅医院产前诊断中心);武泽(云南省第一人民医院生殖医学科);薛晴(北京大学第一医院妇产科);闫丽盈(北京大学第三医院生殖医学科);杨萍(北京大学第三医院血液科);张学(中国医学科学院基础医学研究所医学遗传学系);周灿权(中山大学附属第一医院妇产科);朱小辉(北京大学第三医院生殖医学科);朱依敏(浙江大学医学院附属妇产科医院生殖内分泌科)
所有作者均声明不存在利益冲突

























