
对1例亲代表型正常但生育Dravet综合征患儿的家系进行遗传学分析,通过对一个家庭中表型相似的两个患儿进行全外显子组测序(WES),对患儿及患儿父母进行Sanger测序验证,对患儿父亲精液和外周血DNA突变位点做靶向测序进一步分析,探讨患儿的发病原因。通过WES检测发现患儿SCN1A基因c.5348C>T(p.Ala1783Val)杂合变异,为致病性变异,患儿父亲外周血及精液检查该位点发生嵌合变异,患儿母亲无该位点变异,进而推测父源SCN1A基因嵌合变异是导致子代Dravet综合征发生的遗传学病因。


Dravet综合征又称婴儿严重肌阵挛癫痫综合征,为常染色体显性遗传病。它是一种严重的、难治性的、致残的、多在儿童期发病的发育型癫痫性脑病[1],发病率为1∶15 700~1∶40 900[2],其特征是导致患儿智力障碍和运动控制、行为以及认知障碍。此外,该综合征死亡率高,最常见的死亡原因是癫痫持续状态和癫痫猝死[3]。Dravet综合征是由SCN1A基因变异导致的[4],90%的患儿SCN1A基因变异为新发变异,只有10%来源于父母一方的遗传性变异,父母可为SCN1A杂合变异或者变异嵌合体[5]。亲代变异嵌合体的表型正常或者较轻,嵌合比例与表型之间有一定的相关关系,嵌合比例越高,其临床表型就越严重[6]。本研究对患有Dravet综合征的异卵双胎患儿进行全外显子组测序(WES)及家系的Sanger测序和靶向测序,寻找表型正常的夫妻双方所生育患有Dravet综合征患儿的遗传学病因,为今后的健康生育提供依据和帮助。
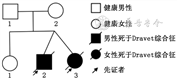
1.病例来源:夫妻双方,结婚9年,孕4产3,7年前顺产1健康女婴,2年前剖宫产异卵双胎1男婴和1女婴,双胎出生后常出现热性惊厥和癫痫持续状态,多数抗癫痫药物治疗无效,两患儿于1岁时夭折,家系图见图1。患儿父母表型正常,无癫痫病史,非近亲结婚。本研究通过郑州大学第一附属医院伦理委员会批准(KY-2021-0102),受检夫妻双方签署知情同意书。
2.DNA提取:在患儿发病期间采集整个家系外周静脉血,采用QIAamp DNA 纯化mini试剂盒(QIAGEN公司,51306)提取基因组DNA。
3. WES:取两患儿基因组DNA,委托北京康旭医学检验所进行WES。采用Agilent Sure Select 外显子组DNA序列富集试剂盒(美国Agilent 公司)制备测序文库,具体操作步骤包括酶切DNA片段、末端修复、磷酸化、加A尾、连接接头、探针杂交、捕获外显子区序列、去除未结合的DNA片段、文库质量检测,应用Illumina测序平台进行WES。
4.Sanger测序变异验证:根据WES检测分析结果,采用Sanger测序对夫妻双方和患儿进行变异验证。针对SCN1A(NM_001202435)基因第26外显子,采用Primer3软件(Primer3webversion4.1.0)设计引物,正向:5′-GCTGCTCTTTGCTTTGATGATG-3′;反向:5′-CACTCTCTCCTAGAACCCGCTT-3′。
5.精液DNA提取:提取采集父亲精液并用QIAamp DNA纯化mini试剂盒(QIAGEN公司,51306)提取精液DNA。
6.靶向测序:根据Sanger测序结果,进一步对父亲精液和外周血DNA变异位点区域PCR片段富集建库进行靶向测序,测序深度为1 000×以上,配合 Illumina 平台测序法,对变异位点进行分析研究。
7.变异位点生物信息学分析:应用UniProt (https://www.uniprot.org/)获取SCN1A的氨基酸序列,运用UCSF Chimera1.14软件在SCN1A编码的蛋白质结构图上进行变异位点标记以及位点变异后所致蛋白结构的变化图。
1.WES结果:两患儿(Ⅱ2与Ⅱ3)SCN1A (NM_001202435)基因第26外显子存在c.5348C>T (p.Ala1783Val)的杂合变异。SIFT和Polyphen2在线软件预测其对蛋白功能影响,结果提示均为有害变异,可能导致蛋白质功能受到影响。根据美国医学遗传学和基因组学学院(ACMG)指南,该变异与先前已确定为致病性的变异有相同的氨基酸改变(PS1),该位点位于热点突变区域(PM1),在ESP数据库、千人数据库、EXAC数据库中正常对照人群中未发现的变异(PM2),且对于SCN1A基因来说,其错义变异是造成特发性癫痫的病因,良性变异比例很小(PP2),用多个软件预测该变异会对基因造成有害影响(PP3),该位点变异被评为致病性变异,与特发性癫痫相关[7]。
2. Sanger测序变异验证:两患儿(Ⅱ2与Ⅱ3)SCN1A基因存在c.5348 C>T(p.Ala1783Val)杂合变异,对父亲(Ⅰ1)外周血DNA测序可见该位点疑似嵌合变异,母亲和表型正常女儿(Ⅰ2与Ⅱ1)均未见变异(图2)。

3.靶向测序:通过测定父亲(Ⅰ1)的精液DNA和外周血进行靶向测序,检测到该位点变异情况和占比,计算父亲精液DNA和外周血DNA的嵌合比例分别为14.25%(12 688/89 037)和6.48%(4 677/72 225)。
4.变异位点生物信息学分析:SCN1A基因第26外显子上c.5348C>T (p.Ala1783Val)变异,使第1 783位丙氨酸变异为缬氨酸,该位点位于蛋白的α亚基的同源区域D4的编码核心区域,通过UCSF Chimera1.14软件分析,该位点与相邻的第1 499位苯丙氨酸产生原子间作用力,影响蛋白质的三级结构,共同导致蛋白质空间结构发生变化(图3),保守性分析该位点在8个不同的物种间完全相同,提示该位点在进化上高度保守。

SCN1A基因编码电压门控钠离子通道α1亚单位,编码蛋白为Nav1.1。哺乳动物的电压门控钠离子通道是由一个α亚基和一个及以上的β亚基构成。α亚基被认为是功能性单位,主要在中枢神经系统表达,β亚基通常被认为是α亚基的辅助性单位,对α亚基的表达起调控作用。α亚基由四个同源结构域组成(D1~D4),每一个结构域有6个α螺旋跨膜片段组成(S1~S6)[8]。S4片段为钠通道的电压感受器,而S5和S6则决定钠通道的选择性和传导性[9]。而与Dravet综合征发生相关的错义变异多发生在编码的核心区(S4~S6),其造成的临床表型较为严重[10]。有研究表明,敲除小鼠的SCN1A基因,导致海马GABA能抑制性中间神经元钠电流显著降低,抑制性中间神经元作用减弱,造成神经系统的兴奋和抑制失衡而导致癫痫发作[11]。
本研究中,两患儿(Ⅱ2与Ⅱ3)SCN1A基因c.5348C>T (p.Ala1783Val)杂合变异,导致编码第1 783位氨基酸由丙氨酸变为缬氨酸,造成相关蛋白的功能受到影响。本家系母亲和表型正常女儿(Ⅰ2与Ⅱ1)的Sanger测序均未发现变异,但是父亲(Ⅰ1)的测序结果提示疑似嵌合,嵌合体可以在体细胞及生殖腺细胞上出现,它是指在同一个受精卵中出现两种或者两种以上的细胞系,这样的嵌合体携带者有可能把变异传递给子代,因此,我们取父亲的精液和外周血DNA做靶向测序进行进一步的验证。靶向测序结果显示精液和外周血DNA均证实父亲(Ⅰ1)为该位点的变异嵌合体,且精液的嵌合比例高于外周血,这与之前Depienne等[6]发现精液的变异比例高于外周血的结论一致,这可能也是导致后代发生Dravet综合征的重要遗传学原因。SCN1A基因位点的变异,不仅会导致后代患Dravet综合征的风险增加,其还与家族性热性惊厥、全面性癫痫伴热性惊厥附加症2型、家族性偏瘫性偏头痛和自闭症等的发生相关[12, 13, 14, 15]。许小菁[16]等通过研究Dravet综合征患儿及亲代临床表型和测序分析得出Dravet综合征患儿SCN1A基因突变率较高,突变类型以错义突变、截断突变为主,只有少数患儿出现SCN1A片段的缺失或重复。有研究表明,95%与Dravet综合征相关的SCN1A基因变异是新生变异,而很少发生在家族性病例中[17],但是这个比例可能比实际偏高,因为在发现新的SCN1A变异之前,一些家族性病例就一直被忽视[18]。Dravet综合征为常染色体显性遗传方式,但是因为父亲的嵌合比例不高,未产生明显的临床表型,在生育患儿之前很难发现其致病性,据报道,76.9%的SCN1A基因变异的嵌合体携带者可以表现出正常或者轻微的症状[19],这在临床上很容易被忽视。所以当正常表型的夫妻生育出患有Dravet综合征的患儿时,夫妻双方一定要进行遗传咨询及变异筛查。朱海燕等[20]对1例亲代表型正常但反复妊娠骨骼发育异常胎儿家系进行分析,在父亲精液DNA检测中发现COL1A2基因存在与胎儿相同位点变异序列,得出父源的COL1A2基因生殖腺嵌合变异是造成胎儿异常的病因。对于表型正常的夫妻生育出异常胎儿要考虑到亲代嵌合体的存在,取材也不能局限于外周血,对生殖细胞的检测同样必要,因为父系或母系嵌合体变异可被下一代遗传,通常分布在患者的所有组织中,包括体细胞和生殖细胞[21]。
本研究对1例父源SCN1A基因嵌合变异导致患儿发生Dravet综合征的家系进行分析,检测出父源SCN1A基因的嵌合体变异,指出嵌合体变异对子代患病的风险的影响,为今后的健康生育和遗传咨询提供重要的理论依据。
所有作者均声明不存在利益冲突

























