
患者男,33岁,慢性病程。临床表现为意外发现右侧肾上腺占位,并行手术切除。随后再次意外发现左侧肾上腺2个占位,手术切除较大占位。随访过程中发现左肾上腺未切除占位增长快速。仔细追问病史,患者生长速度加快时间早,14岁后身高无增加,终身高偏矮(165 cm),提示存在性早熟可能性大,有先天性肾上腺皮质增生症(CAH)的可能。患者病程中血压不高,完善促肾上腺皮质激素、孕酮、17α羟孕酮、中剂量地塞米松抑制试验、基因检测等检查最终明确非经典型21羟化酶缺陷症的诊断。在肾上腺意外瘤患者中,需警惕不典型CAH的可能。对于CAH相关肾上腺占位,无需手术,予糖皮质激素抑制促肾上腺皮质激素可控制肿瘤生长。此外,男性CAH患者易合并下丘脑-垂体-性腺轴功能紊乱,需评估患者性腺轴功能及阴囊B超,以便及时干预。


患者男,33岁,以“双肾上腺占位术后3个月”于2019年4月来北京协和医院内分泌科就诊。患者2010年体检发现右肾上腺占位,直径约4 cm,无不适。就诊于当地医院,相关检验、检查、诊断等不详,行右侧肾上腺肿物切除术,病理检查提示肾上腺皮质腺瘤,大小约3.8 cm×2.5 cm。患者术后无不适,未随诊。2018年11月行肾上腺CT发现左侧肾上腺低密度占位病变,约6.5 cm×3.8 cm×4.9 cm,中等强化(图1A、B);此外,左侧肾上腺内侧近结合部可见稍低密度结节,约2.6 cm×1.6 cm(图1C、D);患者无不适。病程中监测血压波动在120~130/80~90 mmHg(1 mmHg=0.133 kPa)。
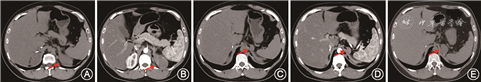
实验室检查:血钠140 mmol/L,血钾4.0 mmol/L,血氯105 mmol/L,葡萄糖4.8 mmol/L。立位醛固酮 267.9 ng/L,立位肾素活性 5.44 ng·ml-1·h-1,醛固酮/肾素活性 4.9,早8点促肾上腺皮质激素(ACTH)为55.2 ng/L,血总皮质醇为76.8 μg/L,24 h尿游离皮质醇 49.20 μg,24 h尿儿茶酚胺未见异常。睾酮、孕酮、17α羟孕酮未查。生长抑素受体显像提示肾上腺占位生长抑素受体高表达,考虑神经内分泌肿瘤可能性大。肾上腺髓质显像未见异常。于2019年1月行左肾上腺较大占位性病变切除,手术保留部分肾上腺,未切除较小占位性病变。术后病理符合肾上腺皮质腺瘤,大小7.5 cm×6.0 cm×3.5 cm。免疫组织化学:Melan-A(+),Vimentin(+),α-inhibin(+),CgA(-),Syn(+),Ki-67(index 10%),CD34(-)。术后规律随诊。2019年4月复查肾上腺CT(图1E),提示未切除的较小病灶较前增大(3.5 cm×2.6 cm),于是进一步就诊于北京协和医院内分泌科。
经询问病史,患者10~11岁出现生长速度加快,14岁后身高无增加,终身高165 cm。既往史无特殊。个人史:28岁结婚,否认勃起功能障碍,诉曾经完善精子活度检测,结果显示“未见异常”,33岁通过辅助生育技术育有两子(双胞胎)。家族史:父亲身高 175 cm,母亲身高 165 cm,通过父母身高预测患者终身高为176.5 cm左右。母亲患有高血压,无肾上腺占位及其他肿瘤家族史。
体格检查:体重 76 kg,身高165 cm,体质指数27.9 kg/m²,血压120/80 mmHg;偏胖体型,皮肤黏膜无明显色素沉着,心率68次/min。心、肺、腹查体未见异常,双侧睾丸体积约18 ml,阴囊及睾丸未触及占位。
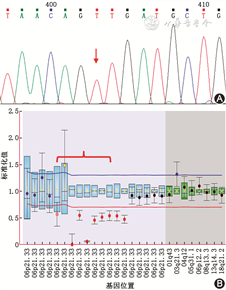
诊治经过:血尿常规、肝肾功能均正常。血钠 138 mmol/L,血钾5.0 mmol/L,血氯 106 mmol/L,葡萄糖4.6 mmol/L。血总皮质醇69 μg/L,24 h尿游离皮质醇 14.8 μg,ACTH 为296.0 ng/L。性激素:促卵泡激素 0.79 U/L,促黄体生成素<0.2 U/L,睾酮 1.12 μg/L,雌二醇 41 ng/L,孕酮 26.46 μg/L,17α羟孕酮 66.27 μg/L,硫酸脱氢表雄酮1 260 μg/L。完善中剂量地塞米松抑制试验(表1),提示孕酮、17α羟孕酮等被明显抑制。LC-MS质谱法类固醇激素测定提示21α羟化酶催化的底物堆积及部分产物减少。阴囊超声:双侧睾丸内部多发低回声,直径多为0.4~0.6 cm,形态规则,边界清,未见血流信号。基因检测提示CYP21A2基因的1条等位基因存在c.518T>A,p.Ile173Asn错义突变,另1条等位基因大片段缺失(图2)。诊断:先天性肾上腺皮质增生症(CAH),21羟化酶缺陷症(非经典型),睾丸肾上腺残基瘤,低促性腺性性功能减退症。治疗:给予泼尼松5 mg 睡前口服。定期复查肾上腺CT,肾上腺占位无进一步增长。
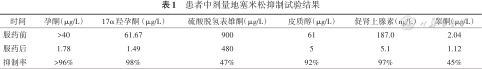
患者中剂量地塞米松抑制试验结果
患者中剂量地塞米松抑制试验结果
| 时间 | 孕酮(μg/L) | 17α羟孕酮 (μg/L) | 硫酸脱氢表雄酮(μg/L) | 皮质醇(μg/L) | 促肾上腺素(ng/L) | 睾酮(μg/L) |
|---|---|---|---|---|---|---|
| 服药前 | >40 | 61.67 | 900 | 61 | 187.0 | 2.04 |
| 服药后 | 1.78 | 1.49 | 480 | 5 | 5.1 | 1.12 |
| 抑制率 | >96% | 98% | 47% | 92% | 97% | 45% |
分析与讨论
本例患者以肾上腺意外瘤起病,最终确诊为21羟化酶缺陷症(非经典型)。肾上腺意外瘤是指非肾上腺相关疾病患者行影像学检查发现的直径>1 cm的肾上腺占位[1]。随着计算机断层扫描、核磁共振的广泛应用,越来越多的肾上腺意外瘤被检出。其检出率随着年龄的增长而增加,平均为4.4%[2],在儿童中少见,在老年人中可高达10%[3]。肾上腺意外瘤的诊治包括评估有无分泌功能及良恶性的鉴别。多数肾上腺意外瘤无功能,约15%的肾上腺意外瘤可分泌过量的激素[1],常见的为皮质醇、醛固酮和儿茶酚胺,分泌雄激素的肿瘤非常罕见。本例患者无典型激素过量分泌的临床表现,曾行皮质醇、醛固酮、儿茶酚胺等激素水平的测定未见异常,考虑患者肾上腺占位无功能。部分肾上腺意外瘤为恶性,包括肾上腺皮质癌及恶性肿瘤肾上腺转移。本例患者肾上腺占位直径>4 cm,肿瘤生长快,不能除外恶性肿瘤的可能,这也是初期外科行手术切除的原因。恶性病变中,肾上腺皮质癌常为单侧肾上腺受累。双侧受累常需要考虑转移瘤、原发性肾上腺淋巴瘤。本例患者为青年男性,未发现其他部位肿瘤,转移瘤和淋巴瘤的可能性不大。对于双侧肾上腺病变,除肿瘤外,还需考虑:肾上腺结核、CAH等[4]。肾上腺结核疾病早期影像学上可表现为双肾上腺增粗,伴或不伴结节,2~3年后常出现钙化。由于肾上腺破坏,患者可出现原发性肾上腺皮质功能减退。病理表现为干酪样病变,与本例病理不符。此外,患者病理结果亦除外了恶性病变的诊断。患者终身高偏矮,通过仔细追问病史,患者10~11岁出现生长速度加快,14岁后身高再无增加,终身高与遗传身高不符,提示性早熟的可能性大,根据这个临床特点,需要怀疑CAH。CAH为常染色体隐性遗传病,致病机制为肾上腺皮质类固醇激素合成过程中某种酶的先天缺陷,下游激素合成不足,ACTH代偿性增多,进而引起肾上腺皮质增生。其中超过95%的CAH为21羟化酶缺陷症[5]。21羟化酶缺陷使孕酮和17羟孕酮不能转化为去氧皮质酮和11去氧皮质醇,造成皮质醇和醛固酮合成障碍,孕酮、17羟孕酮等前体物质产生过多,进一步转化为过多的雄激素。由于该基因的缺陷程度不一,其临床表现差异较大,可分为经典型(包括单纯男性化型、失盐型)和非经典型。严重者可因为盐皮质激素缺乏导致的脱水死亡,典型患者因过量雄激素表现为女性男性化及男性性早熟,轻症患者可无明显症状,仅表现为女性月经不规律及男性终身高偏矮。诊断方面,ACTH兴奋试验是首选的诊断检查,但因国内尚不能获得稳定的ACTH而不能开展。北京协和医院建立中剂量地塞米松抑制试验,当睾酮抑制率>61.2%和(或)17羟孕酮抑制率>87.1%时支持CAH的诊断[6]。基因检测可明确诊断。结合患者临床表现、中剂量地塞米松抑制试验及基因检测,CAH、21羟化酶缺陷症诊断明确。患者病程中无失盐表现,起病隐匿,综合考虑为非经典型21羟化酶缺陷症。
CAH较罕见,临床医师对CAH警惕性不高。
以肾上腺意外瘤起病,肾上腺占位为CAH不典型的影像学表现。
该患者CAH临床表现不典型,初期ACTH升高不明显,极易漏诊。
在肾上腺意外瘤患者中,需警惕不典型CAH的可能。
通过仔细追问病史,完善ACTH、孕酮、17α羟孕酮等检查筛查CAH,必要时完善中剂量地塞米松抑制试验、基因检测明确诊断。
CAH相关的肾上腺占位多不需要手术干预,糖皮质激素可控制其生长。
CAH患者典型肾上腺影像学表现为双侧弥漫性增粗。此外,亦可有其他表现,如结节样增生、肾上腺占位、肾上腺萎缩等,部分患者肾上腺CT可无明显异常。由于过量ACTH长期刺激肾上腺组织,肾上腺占位的患病率高于普通人群[7]。研究报道,CAH患者肾上腺占位直径为1.8~3.5 cm,较大的占位直径可达10 cm以上[7, 8]。半数以上的肾上腺占位为髓样脂肪瘤,其次为肾上腺皮质腺瘤。本患者病理提示肾上腺皮质腺瘤,Ki-67高达10%,这也解释了本例患者占位增长快速的原因。现有文献中鲜有关于皮质腺瘤增殖活性的报道,Chevalier等[9]报道了1例表现为肾上腺占位的CAH患者,病理为肾上腺皮质腺瘤,其Ki-67指数<2%。CAH相关肾上腺占位多无功能,恶性病变的可能性小,无需行手术干预。治疗方面,给予糖皮质激素抑制ACTH即可控制病变进一步生长。也有部分学者提出,对于直径>6 cm的CAH相关肾上腺占位,可考虑行手术切除[9, 10]。
多数21羟化酶缺乏症患者由于雄性激素持续升高,从而导致女性出现男性化,男性出现性早熟。但该患者的睾酮水平偏低。Engels等[11]评估CAH男性患者的性腺功能,发现21羟化酶缺陷患者容易合并下丘脑-垂体-性腺轴功能紊乱。该研究中,20%(14/69)的患者睾酮水平低,其中7例患者为低促性腺性性功能减退,1例患者为高促性腺性性功能减退。本例患者表现为低促性腺性性功能减退症。考虑原因为肾上腺来源的雄激素抑制了性腺轴的功能。此外,睾丸肾上腺残基瘤(TART)压迫睾丸实质,睾丸功能下降引起睾酮水平进一步下降。CAH患者合并TART的概率为27%~49%[11, 12],TART为良性肿瘤,产生的原因是胚胎发育时期,少量肾上腺皮质细胞随睾丸下降,出生后逐渐凋亡,而CAH患者过多的ACTH可刺激残留在睾丸内的肾上腺皮质细胞增殖,逐步增大的肿物可压迫曲细精管等睾丸实质,如果病情得不到控制,可引起睾丸功能下降甚至衰竭。阴囊B超为TART首选的影像学检查,多表现为低回声肿块。该患者阴囊B超提示双侧睾丸多发低回声,TART诊断明确。治疗方面,早期的TART可通过糖皮质激素治疗,肿瘤可以缩小甚至消失,不需要手术干预。女性CAH患者亦可伴发卵巢肾上腺残余肿瘤,临床罕见。
所有作者均声明不存在利益冲突

























