
分析泉州2018—2019年人呼吸道合胞病毒(HRSV)流行情况和基因特征。
样本来自2018年11月至2019年5月福建省泉州市儿童医院送检的下呼吸道感染患儿咽拭子样本,共141份。采用RT-PCR法进行HRSVG基因3′末端靶基因扩增,使用sequencher 5.0以及MEGA5.05软件进行序列编辑、进化树构建和基因特征分析。
HRSV阳性样本25份,17份样本成功获得靶基因序列,其中HRSVA 13份,HRSVB 4份;共鉴定出两个基因型:ON1基因型(13份,HRSVA)和BA9基因型(4份,HRSVB)。5条ON1基因型靶基因序列与2012—2015年北京、长春和浙江流行的ON1基因型序列汇聚为一簇(Cluster1);1条(FJ19-02)与2012—2015年我国多个地区流行的ON1基因型序列汇聚为一簇(Cluster2);其余7条为独立的一簇(Cluster-FJ)。HRSVB亚型中FJ18-02、FJ19-14、FJ19-15与2015年在吉林长春流行的BA9基因型序列汇聚在一起;FJ19-13则与2013年在广州和浙江两地流行的BA9基因型序列具有较近的亲缘性。ON1基因型和BA9基因型均在重复插入的72和60个碱基片段区出现了核苷酸和氨基酸变异。Cluster-FJ分支靶基因序列出现了特征性氨基酸突变(H266L);FJ19-13出现了H261Q和Q265L的独立突变。
2018—2019年福建泉州流行的HRSV基因型为BA9和ON1,Cluster-FJ为新发现的独立传播链,有可能在泉州建立了本土循环流行。


人呼吸道合胞病毒(human respiratory syncytial virus,HRSV)又称人正肺病毒(human orthopneumovirus),为单股、负链、非节段的RNA病毒[1]。HRSV只有一个血清型,又分为A亚型(HRSVA)和B亚型(HRSVB)[2]。根据G基因第二高变区(highly viriable region 2,HVR2)的基因特征[3],将HRSVA分为15个基因型,HRSVB分为26个基因型4, 5, 6]。研究发现HRSV具有A和B亚型交替流行的趋势,2014—2015年ON1基因型逐步取代2008—2013年流行的优势基因型NA1成为我国流行的优势基因型[4];2008—2014年,BA9基因型逐步增多,打破BA基因型与非BA基因型共循环的模式,成为我国流行的HRSVB亚型的优势基因型[7]。目前HRSV呈现全球流行趋势,2015年全球约有3 310万5岁以下儿童由HRSV引起下呼吸道感染[8]。国际并未有安全有效的疫苗和抗体在全球上市以应对HRSV感染。研究发现Nirsevimab单克隆抗体在健康早产儿出生后的第一个HRSV流行季节之前,单次肌肉注射后显著降低了患儿的就诊率和住院率,现已在多个国家开展临床研究[9]。本研究对泉州2018—2019年流行的HRSV进行基因特征分析,为HRSV的防控提供基础数据。
本研究为回顾性分析,收集2018年11月至2019年5月福建省泉州市儿童医院送检的下呼吸道感染患儿咽拭子样本,共141份,所有患儿均在获得其家属知情同意后采集咽拭子样本。
使用QIAamp DNA/RNA mini kit试剂盒(QIAGEN,Valencia,CA,USA,CAT:52906 or 74106)进行病毒总RNA的提取,具体操作参照说明书。使用双通道荧光定量PCR方法进行HRSV样本的鉴定和分型[10];使用一步法逆转录PCR试剂盒One Step RT-PCR kit(TaKaRa Biotechnology Dalian,China,cat:DRR057A)进行HRSV G基因HVR2(634-897 nt)扩增[4],使用ABI Prism3710xl DNA 分析系统进行PCR产物的纯化和测序。
使用sequencher 5.0软件进行序列的编辑整理,下载GeneBank数据库中我国近年流行的HRSV代表株以及HRSV亚型的各基因型定型参考株,使用MEGA5.0软件进行比对,选择邻接法(neighbor-joining method,NJ)和Kimura 2-parameter model替代模型进行亲缘关系进化树的构建以及遗传距离的估算,Bootstrap值选择1 000次重复测试准确性,且Bootstrap>70%以上的数值进行展示[11]。
采用SPSS 17.0软件对患儿基本情况数据进行处理,并进行描述性分析。患儿年龄不符合正态分布,采用M(Q1,Q3)表示。
141例下呼吸道感染患儿年龄为1.0(0.3,4.0)岁,<5岁者占80.1%(113例),<2岁占58.2%(82例);男女比例为1.6/1(87/54)。141份咽拭子样本中,HRSV阳性样本为25份,阳性率为17.7%,其中HRSVA亚型19份,HRSVB亚型6份。共成功扩增17份样本,获得HRSVA HVR2序列13条,2018年1条,2019年12条;HRSVB HVR2序列4条,2018年1条,2019年3条。
13条HRSVA靶基因序列均位于进化树的顶端,与来自加拿大的ON1基因型参考株以及2012—2015年我国流行的ON1基因型序列处于同一分支,因此,该13条HRSVA靶基因序列鉴定为ON1基因型。其中FJ18-01、FJ19-03、FJ19-05、FJ19-06、FJ19-07与2012—2015年在我国北京、长春、浙江等地流行的ON1基因型序列汇聚为一簇(Cluster 1);FJ19-02与2012—2015年我国北京、长春、湖南、陕西等地流行ON1基因型序列汇聚为一簇(Cluster 2);余下7条序列(FJ9-01、FJ19-04、FJ19-08、FJ19-09、FJ19-10、FJ19-11、FJ19-12)汇聚为独立的一簇(Cluster-FJ),未与2012—2015年我国流行的HRSVA ON1基因型序列汇聚在一起(图1A)。
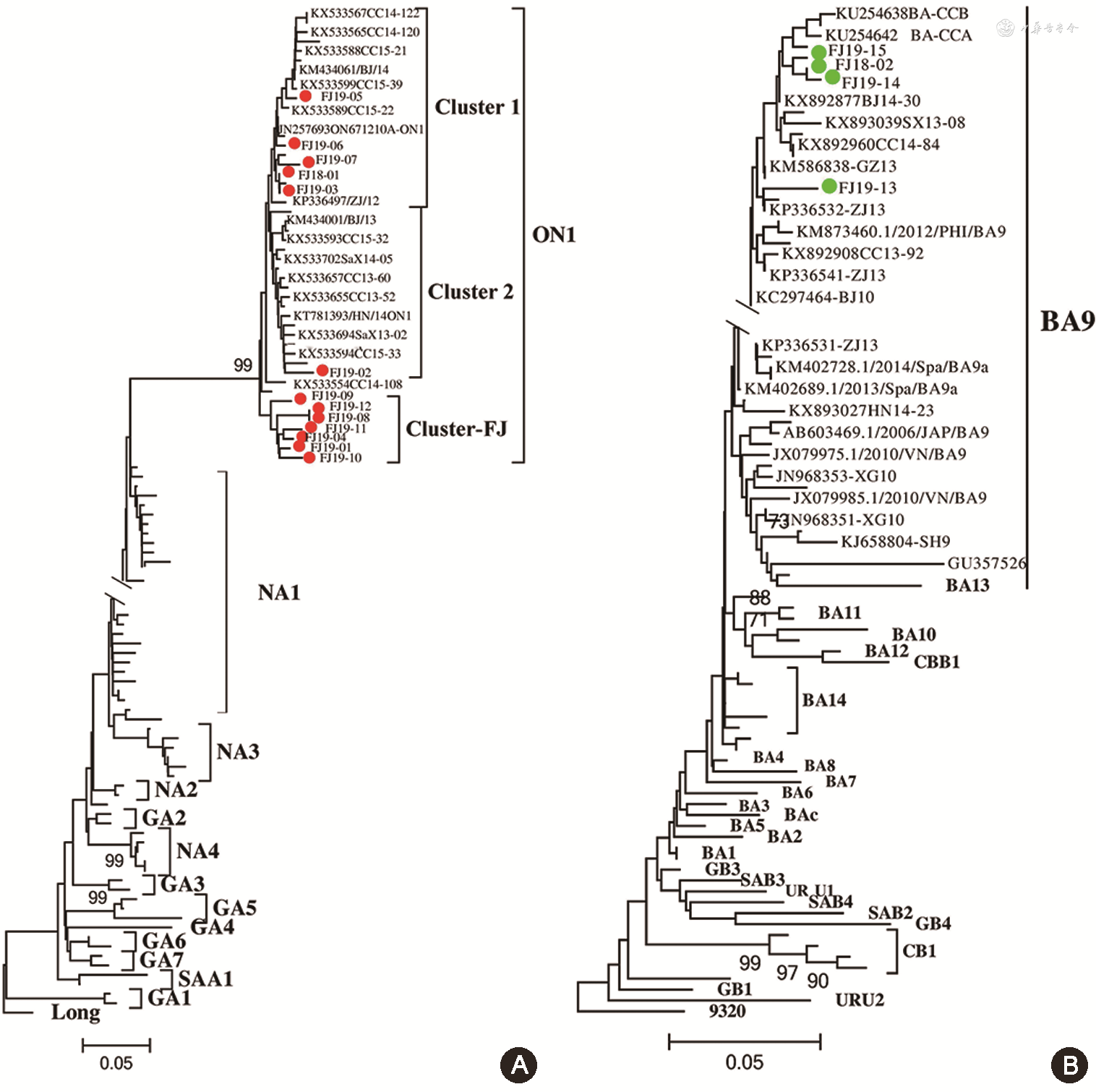
4条HRSVB G基因HVR2区核苷酸序列分散在2008—2015年在中国浙江、吉林、北京、广州、陕西等地流行的BA9基因型序列,以及2006年至今来自全球多个国家流行的BA9基因型参考株汇聚成的分支上,故此4条HRSVB亚型靶基因序列鉴定为BA9基因型。其中FJ18-02、FJ19-14、FJ19-15与2015年在我国吉林长春流行的BA9基因型序列具有较近的亲缘关系,与2013—2016年在我国北京、陕西、长春等地流行的BA9基因型序列汇聚成一个分支,位于树的顶端;FJ19-13则与2013年在我国广州和浙江流行的BA9基因型序列汇聚在一起(图1B)。
同参考株(GenBank号:JN257694)比较,13条ON1基因型HRSVA序列的核苷酸变异个数分别为4个(FJ19-06)、5个(FJ19-05和FJ18-01)、7个(FJ19-07)、8个(FJ19-03和FJ19-02)、9个(FJ19-01)、10个(FJ19-09和FJ19-04)、11个(FJ19-11)、14个(FJ19-10、FJ19-12和FJ19-08),核苷酸序列变异率在1.2%~4.2%。进一步对其编码氨基酸的差异分析表明,与NA1基因型相比,ON1基因型在氨基酸位点283位后出现了72个核苷酸(24个氨基酸)的插入,且插入片段C2-C24与aa261-283氨基酸序列重复。ON1基因型除与NA1基因型共出现的15个氨基酸位点突变外,ON1基因型参考株及HRSVA FJ序列在氨基酸位点aa232、aa253和aa273出现特有突变,且7条序列(FJ9-01、19-04、19-08、19-09、19-10、19-11、19-12)由于在核苷酸797位的A797T突变导致氨基酸位点现H266L的有义突变(图2A)。
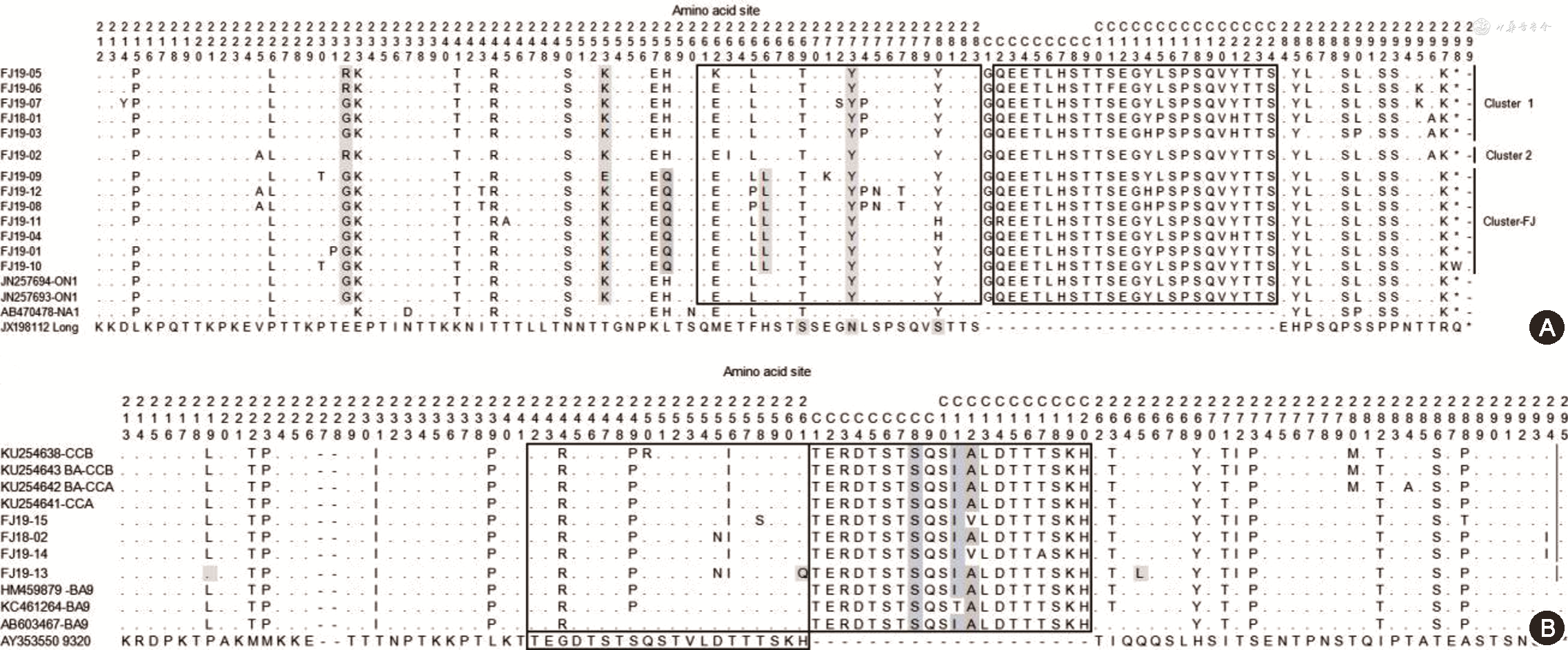
与BA9基因型参考株(GenBank号:AB603467)比较分析,4条HRSVB亚型靶基因核苷酸序列变异个数分别为11和12个(FJ19-13),核苷酸变异率在3.6%和4.0%。BA基因型为1998年发现的在氨基酸位点283后出现20个氨基酸的重复插入,该插入片段并非与原片段(aa242-aa261)完全重复,BA9基因型在插入的20个氨基酸中的第8位与原型株9320一致(S),与BA9基因型的aa249位(P)不同;插入片段的第11位(I)与原型株及BA9基因型的aa252不同(T);FJ19-14和FJ19-15插入片段的第12位为A,而FJ18-02和FJ19-13与原型株及BA9基因型aa253位点一致(V)。FJ19-13和原型株在aa219位保持一致,在aa261位点出现H261Q的突变,在aa265出现Q265L的突变(图2B)。
HRSV是引起婴幼儿以及老年人下呼吸道感染的常见病原体之一。我国急性下呼吸道感染住院患者病原构成中,病毒性病原检出率为36.6%,其中HRSV检出率为9.9%[12],而且HRSV在幼年组检出率较高,易出现混合感染,检出高峰为冬春季[13, 14]。分子流行病学研究是基于PCR技术研究不同年代地域病毒的某一段基因的变化特征,可以为诊断以及病毒病的防控提供基础,比如针对目前仍然在流行的新型冠状病毒,通过对目的基因的鉴定,能够快速地进行核酸检测从而辅助诊断[15],而目前大部分疫苗的开发与评价,仍然基于病毒的基因组特征[16],如果某一关键位点出现突变,那么就有可能影响到疫苗的保护效果,所以分子流行病学研究通过对目前流行病毒的基因特征分析,为病毒学研究提供基础数据,是分子诊断、疫苗和抗体研发的基础,同时也能为疫苗分子水平评价和监控提供技术平台。
本研究发现,2018—2019年流行于泉州的HRSVA亚型序列均为ON1基因型,说明ON1基因型病毒已在福建泉州地区建立了循环流行。本研究佐证了ON1基因型确实已取代NA1基因型成为我国流行的优势基因型并持续传播[4]。Cluster-FJ在亲缘性关系树上形成独立一簇,提示泉州流行的ON1独立于其他地区流行的HRSV病毒,但由于缺乏其他地区的病毒学监测数据和相关的流行病学信息,尚不能鉴定其传播来源。有必要对泉州和我国其他地区流行的HRSV病毒进行连续监测,以明确该新发现的Cluster-FJ 分支病毒在泉州和其他地区的传播和流行。
本课题组在既往研究中发现BA9基因型自2006年在我国出现广泛流行,但并未出现明显的基因型更替以及进化特征[7]。但是本研究中来自泉州的3条序列与长春2015年流行的BA9基因型序列汇聚在树的顶端,推测该簇病毒在流行过程中出现了病毒进化,而FJ19-13 HRSV仅与浙江地区流行的BA9基因型病毒处于同一进化分支。说明BA9基因型在我国广泛传播的同时,又有不同的小分支在局部地区流行。
文献研究揭示B亚型的进化速率略高于A亚型[17],本研究发现BA9基因型的变异率也较ON1基因型的变异率稍高。ON1基因型Cluster-FJ病毒出现H266L的突变,BA9基因型序列FJ19-13也出现H261Q和Q265L的特有突变,除H261Q突变仅改变支链所带电荷的性质外,其余2个氨基酸的突变均出现氨基酸亲疏水性以及极性的改变,这些突变位点是否影响了G蛋白的结构、增加了病毒的黏附能力、影响了病毒功能,是否是导致Cluster-FJ分支病毒流行传播的原因等科学问题尚需进一步研究。文献研究表明与无60个核苷酸插入片段的BA基因型病毒相比,有插入片段的BA基因型病毒能够增加病毒的黏附以及适应能力,从而有利于病毒的感染和传播[18]。那么ON1基因型出现的大片段的插入是否具有类似的功能还需要进一步研究。
综上,BA9和ON1基因型是2018—2019年泉州流行的基因型,且存在不同的传播链。Cluster-FJ,是新发现的独立传播链,引起了福建泉州局部地区的小流行。但由于监测时间较短和样本数量较少的局限性,尚需要进行连续监测以获得HRSV流行特征和病毒变异变迁规律,为HRSV的诊断、单克隆抗体及药物和疫苗的研制提供基础数据。
所有作者均声明不存在利益冲突

























