
分析不同基因型Prader-Willi综合征(PWS)患者临床生化特征和治疗效果的差异。
回顾性纳入2017年5月至2018年12月于北京协和医院内分泌科门诊就诊的35例PWS患者,其中男20例,女15例,年龄[M(Q1,Q3)][3.0(0.8,10.0)]岁。收集患者的临床和生化资料,并采集外周血标本。提取患者外周血白细胞DNA,用甲基化特异性多重连接探针扩增技术(MS-MLPA)检测患者基因缺失或甲基化异常,并据此分组(父源缺失组27例,甲基化异常组8例),分析两组患者生化检查结果及生长激素的治疗效果。
MS-MLPA技术检测示77%(27/35)的患者基因型为父源缺失型,23%(8/35)患者为甲基化异常。生化检查结果方面,父源缺失组患者尿酸(UA)水平高于甲基化异常组[(363±101)μmol/L比(259±74)μmol/L,P=0.019]。体重与尿酸水平之间存在线性关系,经体重校正后,父源缺失组与甲基化异常组患者尿酸水平差异无统计学意义(P=0.101)。两组患者均应用生长激素[剂量:每天(0.14±0.03)U/kg]治疗。父源缺失组患者随访(26.0±13.6)个月,身高由(99.0±31.5)cm[(-0.3±1.1)同年龄同性别儿童身高标准差(SDS)]增加至(107.5±27.0)cm[(0.7±0.9)SDS](P=0.037);甲基化异常组患者随访(25.8±11.6)个月,患者身高由(86.4±31.2)cm[(-0.7±1.8)SDS]增加至(95.6±26.5)cm[(0.0±1.6)SDS](P=0.557)。治疗前后父源缺失组和甲基化异常组患者体质指数差异无统计学意义[(22.0±7.1)kg/m2比(22.4±6.8)kg/m2,P=0.890;(17.0±3.1)kg/m2比(16.4±2.7)kg/m2,P=0.754]。
父源缺失和甲基化异常的PWS患者生化检查结果差异无统计学意义。PWS患者早期应用生长激素治疗可有效促进身高增长,控制体重增加。


Prader-Willi综合征(PWS)又称普拉德-威利综合征,其发病率为1/10 000~1/30 000[1]。该类患者在新生儿期和婴儿期主要表现为肌张力低下和喂养困难,儿童期和成年期主要表现为过度摄食而导致肥胖,同时可有特征性面容、发育迟缓、性腺功能低下、身材矮小等[1]。PWS是由于父源染色体15q11.2~q13区域印记基因功能缺陷所导致的遗传性疾病[2],主要遗传类型包括3种:父源缺失型、母源同源二倍体(UPD)和印记中心微缺失及突变[2]。不同基因分型患者对生长激素治疗反应有所不同。因此,明确PWS患者的基因分型对患者治疗效果及预后有一定指导意义。本研究旨在通过明确PWS患者遗传类型,比较分析不同遗传类型患者的生化检测结果及生长激素的治疗效果,以提高对此类疾病的认识和诊治水平。
本研究为病例系列分析,回顾性纳入2017年5月至2018年12月于北京协和医院内分泌科门诊就诊的35例PWS患者,男20例,女15例,年龄3.0(0.8,10.0)岁。PWS患者诊断标准主要参考《中国Prader-Willi综合征诊治专家共识》的临床评分标准[3],包括6条主要标准,11条次要标准和8条支持证据。年龄<3岁总评分5分以上,主要诊断标准达4分即可诊断;年龄≥3岁,总评分8分以上,主要标准达5分即可诊断。本研究患者纳入标准:(1)临床特征符合PWS诊断标准;(2)未接受生长激素治疗;(3)规律随访半年及以上的患者纳入治疗随访分析。排除标准:(1)初诊时已接受生长激素治疗;(2)临床资料缺失或无法规律随访的患者;(3)未行基因诊断的患者。经患者及其家长同意并签署知情同意书后,收集患者外周血,进行基因水平的研究。本研究通过北京协和医院伦理委员会审核(JS-2111)。
1. 临床资料收集:收集患者的一般临床资料、体格检查和实验室检查结果(部分患者临床资料缺失)。
2. 血标本采集和DNA提取:每例患者采集2 ml外周血,按血液DNA提取试剂盒(Omega Blood DNA Midi Kit,美国Omega Biotek公司)说明书的标准流程提取DNA并测定浓度及纯度。
3. 甲基化特异性多重连接探针扩增技术(MS-MLPA)检测患者基因缺失或甲基化异常:采用MS-MLPA试剂盒(荷兰MRC-Holland公司)进行检测。该试剂盒在染色体15qll~q13的PWS/AS关键区域有34对特异性探针,其中5个针对印迹序列的探针包含有限制性内切酶Hha I的识别位点,用于判断该序列的甲基化状态。实验流程根据试剂盒说明进行,主要步骤包括:DNA变性、分子杂交、连接反应和特异PCR扩增。其中在进行连接反应时,DNA样品被平均地分为两部分,一部分进行单纯的连接反应,另一部分加入甲基化敏感的Hha I酶切,使连接和酶切反应同时进行。扩增后的PCR产物用测序仪ABI 3710进行基因型分析,最终结果使用Coffalyzer软件(荷兰MRC-Holland公司)进行分析。每个实验纳入3个正常对照样本,通过比较目标样本与正常对照样本的峰值,判断基因拷贝数和甲基化变异情况。
由于MS-MLPA无法区分UPD和印记中心甲基化异常,因此根据检测结果,将患者分为父源缺失组及甲基化异常组,其中父源缺失组27例,甲基化异常组8例。
采用SPSS 21.0软件对数据进行分析。游离三碘甲状腺原氨酸(FT3)、游离甲状腺素(FT4)、睾酮(T)、雌二醇(E2)、8点血皮质醇(F8 am)、低密度脂蛋白胆固醇(LDL-c)、尿酸符合正态分布,采用表示,两组比较采用独立样本t检验。年龄、身高、体重、胰岛素样生长因子1(IGF1)、促甲状腺激素(TSH)、黄体生成素(LH)、卵泡刺激素(FSH)、空腹胰岛素(FINS)、空腹血糖(Glu)、总胆固醇(TC)、甘油三酯(TG)、高密度脂蛋白胆固醇(HDL-c)为偏态分布,以M(Q1,Q3)表示,采用Wilcoxon秩和检验比较两组差异。采用率和频数描述性别、出生产式、隐睾发生率、基因型等定性资料。根据基因型将患者分为父源缺失组和甲基化异常组,以尿酸为因变量,分组为固定因子,体重为协变量进行协方差分析比较体重对尿酸的影响。双侧检验,检验水准α=0.05。
35例PWS患者的身高为94.0(70.0,137.4)cm,体重为19.8(9.2,48.6)kg。80%(20/25)患者出生为剖宫产,20%(5/25)为自然分娩。20例男性患者中,18例合并隐睾;此外,2例患者合并脊柱侧弯,各有1例患者合并足内翻、双肾积水或脐疝。59%(20/34例)的患者IGF1低于正常水平,其中14例患者基因型为父源缺失,6例患者为甲基化异常。所有骨龄>12岁或生物年龄>18岁男性及生物年龄>14岁[4]的女性患者均存在性腺功能减退症(共6例)。6%(2/33例)的患者存在继发性甲状腺功能减退症,表现为FT4水平低于正常(正常范围:8.88~10.42 pmol/L),但TSH水平降低或处于正常范围(正常范围:0.30~2.07 U/L)。
利用MS-MLPA技术,在35例PWS患者中检测到28例患者存在15q11~q13的片段缺失,即父源缺失型,其中,表现为Ⅰ型缺失的患者为7例,Ⅱ型缺失的患者为20例,其余8例患者存在甲基化异常(图1)。
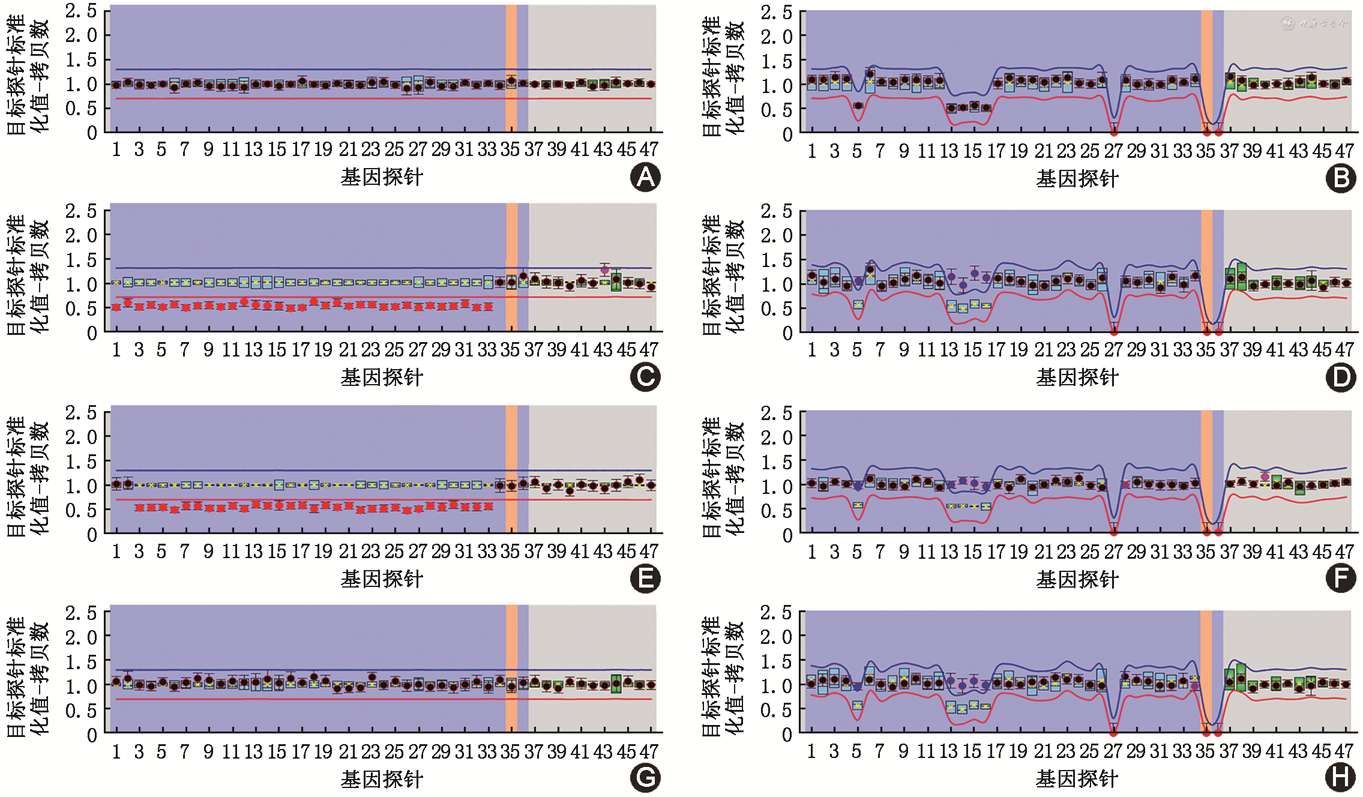
注:横坐标为基因探针(1为TUBGCP5-8;2为NIPA1-3;3为MKRN3-1;4为MAGEL2-1;5为MAGEL2-1;6为NDN-1;7为SNRPN-u1b;8为SNRPN-u1b;9为SNRPN-Intr.u2;10为SNRPN-Intr.u2;11为SNRPN-u5;12为SNRPN-u5;13为SNRPN-5;14为SNRPN-3;15为SNRPN-3;16为SNRPN-3;17为SNRPN-3;18为SNRPN-7;19为SNRPN-HB2-85-SNORD;20为SNRPN-HB2-85-SNORD;21为SNRPN-HB2-85-SNORD;22为UBE3A-9;23为UBE3A-4;24为UBE3A-3;25为UBE3A-2;26为UBE3A-1;27为UBE3A-1;28为ATP10A-15;29为ATP10A-1;30为GABRB3-9;31为GABRB3-7;32为OCA2-23;33为OCA2-3;34为APBA2-14;35为SLC9A2-2;36为ITSN1-12;37~47为参考探针)。浅蓝色方框代表通过对照样本所绘制的该探针拷贝数或甲基化的95%CI;实心圆代表目的探针,上下端线为该探针拷贝数或甲基化的最高和最低范围;平行红色及蓝色线条代表探针的拷贝数或甲基化的边界,高于此边界表示拷贝数或甲基化增多,低于此边界表示拷贝数或甲基化减少
将35例患者根据基因型进行分组,父源缺失组患者尿酸水平高于甲基化异常组尿酸水平[(363±101)μmol/L比(259±74)μmol/L](t=2.499,P=0.019)。两组其余指标(TSH、FT3、FT4、IGF1、LH、FSH、T、E2、F8 am、Glu、FINS、TC、TG、HDL-c、LDL-c)水平差异均无统计学意义(均P>0.05)(表1)。进一步行协方差分析,结果显示体重与尿酸水平之间存在线性关系,经体重校正后,父源缺失组与甲基化异常组患者尿酸水平差异无统计学意义(F=2.893,P=0.101)。
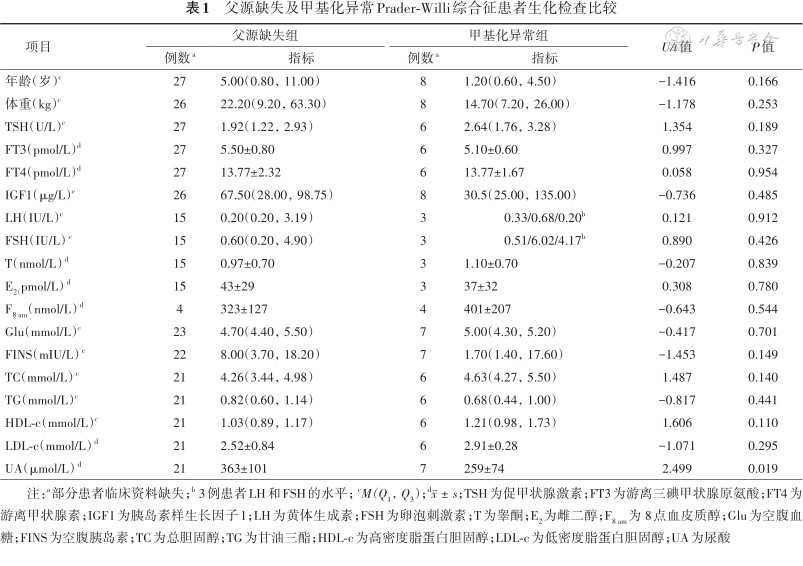
父源缺失及甲基化异常Prader-Willi综合征患者生化检查比较
父源缺失及甲基化异常Prader-Willi综合征患者生化检查比较
| 项目 | 父源缺失组 | 甲基化异常组 | U/t值 | P值 | ||
|---|---|---|---|---|---|---|
| 例数a | 指标 | 例数a | 指标 | |||
| 年龄(岁)c | 27 | 5.00(0.80,11.00) | 8 | 1.20(0.60,4.50) | -1.416 | 0.166 |
| 体重(kg)c | 26 | 22.20(9.20,63.30) | 8 | 14.70(7.20,26.00) | -1.178 | 0.253 |
| TSH(U/L)c | 27 | 1.92(1.22,2.93) | 6 | 2.64(1.76,3.28) | 1.354 | 0.189 |
| FT3(pmol/L)d | 27 | 5.50±0.80 | 6 | 5.10±0.60 | 0.997 | 0.327 |
| FT4(pmol/L)d | 27 | 13.77±2.32 | 6 | 13.77±1.67 | 0.058 | 0.954 |
| IGF1(μg/L)c | 26 | 67.50(28.00,98.75) | 8 | 30.5(25.00,135.00) | -0.736 | 0.485 |
| LH(IU/L)c | 15 | 0.20(0.20,3.19) | 3 | 0.33/0.68/0.20b | 0.121 | 0.912 |
| FSH(IU/L)c | 15 | 0.60(0.20,4.90) | 3 | 0.51/6.02/4.17b | 0.890 | 0.426 |
| T(nmol/L)d | 15 | 0.97±0.70 | 3 | 1.10±0.70 | -0.207 | 0.839 |
| E2(pmol/L)d | 15 | 43±29 | 3 | 37±32 | 0.308 | 0.780 |
| F8 am(nmol/L)d | 4 | 323±127 | 4 | 401±207 | -0.643 | 0.544 |
| Glu(mmol/L)c | 23 | 4.70(4.40,5.50) | 7 | 5.00(4.30,5.20) | -0.417 | 0.701 |
| FINS(mIU/L)c | 22 | 8.00(3.70,18.20) | 7 | 1.70(1.40,17.60) | -1.453 | 0.149 |
| TC(mmol/L)c | 21 | 4.26(3.44,4.98) | 6 | 4.63(4.27,5.50) | 1.487 | 0.140 |
| TG(mmol/L)c | 21 | 0.82(0.60,1.14) | 6 | 0.68(0.44,1.00) | -0.817 | 0.441 |
| HDL-c(mmol/L)c | 21 | 1.03(0.89,1.17) | 6 | 1.21(0.98,1.73) | 1.606 | 0.110 |
| LDL-c(mmol/L)d | 21 | 2.52±0.84 | 6 | 2.91±0.28 | -1.071 | 0.295 |
| UA(μmol/L)d | 21 | 363±101 | 7 | 259±74 | 2.499 | 0.019 |
注:a部分患者临床资料缺失;b 3例患者LH和FSH的水平;cM(Q1,Q3);d;TSH为促甲状腺激素;FT3为游离三碘甲状腺原氨酸;FT4为游离甲状腺素;IGF1为胰岛素样生长因子1;LH为黄体生成素;FSH为卵泡刺激素;T为睾酮;E2为雌二醇;F8 am为 8点血皮质醇;Glu为空腹血糖;FINS为空腹胰岛素;TC为总胆固醇;TG为甘油三酯;HDL-c为高密度脂蛋白胆固醇;LDL-c为低密度脂蛋白胆固醇;UA为尿酸
35例PWS患者中16例患者进行了规律随访,其中父源缺失组11例,UPD组5例。两组患者生长激素用量均为每天(0.14±0.03)U/kg,随访时间分别为(26.0±13.6)个月和(25.8±11.6)个月。治疗后,父源缺失组患者身高由(99.0±31.5)cm[(-0.3±1.1)同年龄同性别儿童身高标准差(SDS)]增加至(107.5±27.0)cm[(0.7±0.9)SDS],甲基化异常组患者身高由(86.4±31.2)cm[(-0.7±1.8)SDS]增加至(95.6±26.5)cm[(0.0±1.6)SDS]。治疗前后两组患者体质指数(BMI)差异无统计学意义(t=-0.140、0.324,P>0.05)(表2)。
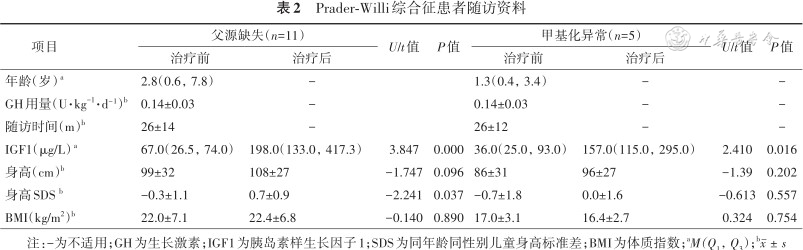
Prader-Willi综合征患者随访资料
Prader-Willi综合征患者随访资料
| 项目 | 父源缺失(n=11) | U/t值 | P值 | 甲基化异常(n=5) | U/t值 | P值 | ||
|---|---|---|---|---|---|---|---|---|
| 治疗前 | 治疗后 | 治疗前 | 治疗后 | |||||
| 年龄(岁)a | 2.8(0.6,7.8) | - | 1.3(0.4,3.4) | - | - | |||
| GH用量(U·kg-1·d⁻¹)b | 0.14±0.03 | - | 0.14±0.03 | - | - | |||
| 随访时间(m)b | 26±14 | - | 26±12 | - | - | |||
| IGF1(μg/L)a | 67.0(26.5,74.0) | 198.0(133.0,417.3) | 3.847 | 0.000 | 36.0(25.0,93.0) | 157.0(115.0,295.0) | 2.410 | 0.016 |
| 身高(cm)b | 99±32 | 108±27 | -1.747 | 0.096 | 86±31 | 96±27 | -1.39 | 0.202 |
| 身高SDS b | -0.3±1.1 | 0.7±0.9 | -2.241 | 0.037 | -0.7±1.8 | 0.0±1.6 | -0.613 | 0.557 |
| BMI(kg/m2)b | 22.0±7.1 | 22.4±6.8 | -0.140 | 0.890 | 17.0±3.1 | 16.4±2.7 | 0.324 | 0.754 |
注:-为不适用;GH为生长激素;IGF1为胰岛素样生长因子1;SDS为同年龄同性别儿童身高标准差;BMI为体质指数;aM(Q1,Q3);b
PWS是基因组印记相关的遗传性疾病的典型代表,确诊需要依赖基因检测手段。诊断方法包括甲基化特异性PCR(MS-PCR)、荧光原位杂交(FISH)、微阵列基因分析(CMA)、MS-MLPA等。MS-MLPA已有配套试剂,是目前常用的诊断方法[5]。其可以同时检测15号染色体12个基因缺失、重复突变,结果符合率≥99%。本研究结果显示80%的PWS患者为父源缺失型,20%的患者为甲基化异常,与既往文献报道结果相似[6]。本研究示父源缺失组患者尿酸水平高于甲基化异常组患者,差异有统计学意义。尿酸是嘌呤代谢的终末产物,与饮食、年龄、肥胖等密切相关[7]。父源缺失组患者年龄相对较大,且体重高于甲基化异常组患者,经体重校正后两组患者尿酸水平无差异,说明肥胖所致代谢紊乱可能是其尿酸水平高的主要影响因素。
生长激素缺乏症是PWS患者较为常见的内分泌异常,发生率为40%~100%[8, 9]。PWS患者的甲状腺功能减退多为继发性,发生率为2%~30%[10, 11]。本研究中59%的患者存在生长激素缺乏症,6%的患者存在继发性甲状腺功能先退,与既往文献报道一致。研究显示绝大多数 PWS患者存在不同程度的性腺功能减退,包括低促性腺激素性性腺功能减退症(HH)或原发的性腺功能减退。男性PWS患者隐睾发生率高达66%~100%[8]。本研究中90%男性患者合并隐睾,且100%患者存在性腺功能减退症,与既往研究报道一致。由于存在下丘脑功能障碍,PWS患者存在中枢性肾上腺皮质功能不全(CAI)的可能,发生率为0~60%[12, 13, 14]。本研究患者的血皮质醇均处于正常范围,临床无肾上腺皮质功能减退的相关表现,提示患者CAI可能性小,但未行相关刺激试验无法准确评估患者是否存在CAI。
生长激素替代治疗除了改善PWS患者身高外,还可降低脂肪含量,增加瘦体重。本研究随访的PWS患者年龄为0.3~11.5岁,随访26个月后父源缺失组患者及甲基化异常组患者身高均有明显增加,而BMI无明显增加,提示早期应用生长激素可以有效改善PWS患者身高,控制体重快速增长。不同基因型的PWS患者对生长激素治疗效果可能不同。父源缺失型患者的瘦体重增加可能优于UPD患者,而UPD患者身高增长则可能优于父源缺失患者[15]。本文结果显示生长激素治疗后父源缺失组患者身高增加1 SDS,甲基化异常组患者身高增加0.7 SDS,提示父源缺失组患者身高改善可能优于甲基化异常组患者。目前关于生长激素治疗对不同基因型PWS治疗疗效的研究结论尚不明确,需大样本量研究进一步确证。此外,研究发现早期应用生长激素治疗可以促进PWS患儿精神运动发育,提高认知能力[16]。本研究中心在随访PWS患者过程中也观察到早期应用生长激素治疗患者的反应能力、运动协调性、语言能力等方面均有改善,提示生长激素治疗对神经系统发育有一定作用。但因本研究为回顾性研究,未能纳入客观性指标评估患者神经系统发育,为其局限性。
综上所述,本研究利用MS-MLPA技术明确35例PWS患者的基因类型,为临床精准诊疗提供了基础数据资料。PWS患者常合并多种内分泌异常,包括性腺功能减退、生长激素缺乏及甲状腺功能减退等。父源缺失和甲基化异常的PWS患者内分泌激素检查结果无差异。生长激素治疗可有效改善PWS患者的身高,父源缺失组患者对生长激素治疗反应可能优于甲基化异常组患者。
所有作者均声明不存在利益冲突

























