
Castleman病是一类罕见的血液系统疾病。本文报道1例以膜性肾病、多浆膜腔积液为主要临床表现的不典型的Castleman病患者,病程中合并反复肾周出血,病情迁延复杂,经多学科协作明确诊断并给予有效治疗。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者男,36岁,2021年5月27日因“水肿9年,腰痛7年,腹围增大1年”收入北京协和医院消化科。2012年2月患者出现全身可凹性水肿,尿中泡沫增多,血白蛋白(Alb)21.5 g/L,血肌酐(SCr)62.3 μmol/L,24 h尿蛋白3 g。超声:双肾增大并弥漫性病变。肾脏穿刺病理:膜性肾病Ⅲ期。外院予泼尼松60 mg/次、1 次/d及环磷酰胺治疗,用药2个月后水肿消失,复查血生化、尿常规无异常,2014年2月停药。2014年4月患者出现右侧腰痛,血红蛋白(Hb)147 g/L,SCr 102 μmol/L,CT:右肾周弧形高密度影,考虑被膜下血肿;双肾盂积水;腹腔积液。外院予对症治疗后出院。2020年初腹围增加,肚脐膨出。近1年3次突发左侧剧烈腰痛,伴恶心、呕吐胃内容物,CT提示左肾包膜下血肿可能,腹腔积液。2020年12月外院行腹腔穿刺,引流淡红色血性腹水,约24 h后逐渐转为无色透明腹水。末次就诊行正电子发射型计算机断层扫描/磁共振成像(PET/MRI):双肾包膜下、双侧肾盂旁积液,代谢不高;左侧胸腔积液,腹水。肾动脉核磁共振血管造影(MRA):双肾动脉主干及一级分支显影浅淡,二级分支显影欠清。腰痛发作时予对症治疗,持续1~2 d好转。因双肾积水可能,置入左侧输尿管支架管(D-J管)。近1个月腹胀明显,腹围由95 cm增至103 cm,平卧位喘憋、干咳,坐位缓解,伴活动耐量下降。病程中否认特殊药物服用史,否认饮酒史。入院查体:体温 36.5 ℃,呼吸17次/min,脉搏131次/min,血压109/66 mmHg(1 mmHg=0.133 kPa),不吸氧状态下动脉血氧饱和度90%,腹围103 cm,体重66 kg。颈部触及多枚肿大淋巴结,质韧,最大者直径约2 cm。心律齐,心率快。双下肺呼吸音低。腹部膨隆,可见脐疝及多发宽大紫纹,腹软,无压痛、反跳痛、肌紧张。液波震颤(+)。
入院后实验室检查:Hb 134 g/L,白细胞(WBC)11.42×109/L,血小板528×109/L。血生化:Alb 29 g/L,丙氨酸转氨酶 106 U/L,天冬氨酸转氨酶 60 U/L,碱性磷酸酶677 U/L,谷氨酰胺转移酶 334 U/L,总胆红素12.4 μmol/L,磷1.8 mmol/L,钙2.24 mmol/L,SCr 138 μmol/L。超敏C反应蛋白(hsCRP)>250 mg/L,红细胞沉降率(ESR)86 mm/1h,白细胞介素(IL)-6 104.0 ng/L,IL-8 68.0 ng/L,肿瘤坏死因子(TNF)-α 17.3 ng/L,血管内皮生长因子(VEGF)270.0 ng/L。外周血抗PLA2R:阴性。血清蛋白电泳、免疫固定电泳、自身免疫性疾病及感染指标均阴性。24 h尿蛋白0.4 g。腹水常规:WBC 92×106/L,单核细胞 73×106/L,黎氏试验(-)。腹水生化:总蛋白 5 g/L,Alb 4 g/L(同期血清Alb 30 g/L),腺苷脱氢酶 0.6 U/L,乳酸脱氢酶 44 U/L,葡萄糖 5.0 mmol/L。病原学、肿瘤标志物、肿瘤细胞:阴性。超声:双侧锁骨上窝及颈部多发肿大淋巴结,皮髓质分布不清,较大者位于右侧锁骨上窝(1.9 cm×1.3 cm)。泌尿系超声:双肾盂无回声,内见分隔,符合肾盂周囊肿。心脏超声:未见明显异常。CT尿路造影:双肾盂周围低密度灶,肾盂周囊肿可能;双肾体积增大,密度、强化不均,双肾周可见积液包绕;左侧D-J管置入术后;多浆膜腔积液;门静脉系统未见血栓(图1)。淋巴显像:肾实质弥漫性淋巴管扩张;双下肢内侧集合淋巴管、左静脉角淋巴管扩张可能;双肾盂囊肿占位,肾周低密度影,腹腔见大量积液。尿乳糜试验、腹水乳糜试验均阴性。肾脏穿刺病理本院会诊:符合膜性肾病,合并肾小管间质病变。病理结果:(右侧锁骨上淋巴结)符合Castleman病(透明血管型)(图2)。因患者尿蛋白量少,提示膜性肾病非复发状态,膜性肾病无法解释患者反复肾周出血、双肾盂周囊肿,且患者合并大量腹水,行肾脏穿刺难度大、出血风险高,故未行重复肾活检。治疗方面予间断引流腹水(共计约12 000 ml)、补充白蛋白、呋塞米+螺内酯联合利尿,胸闷、憋气、干咳症状好转,拔除引流管,体重稳定于55 kg左右,腹围82 cm,脉搏70~80 次/min,不吸氧状态下动脉血氧饱和度96 %。复查hsCRP 5.7 mg/L,IL-6 15.4 ng/L,IL-8 16.0 ng/L,IL-10 5.0 ng/L,TNF-α 15.9 ng/L,血清Alb 36 g/L,SCr 101 μmol/L,胸腹盆CT示胸水及腹水明显减少。经内科大查房(消化科、肾内科、血液科、放射科、核医学科、病理科)讨论,考虑特发性多中心Castleman病(iMCD)诊断明确,目前已无疾病活动证据,建议随诊观察,若再出现疾病活动表现,可针对iMCD治疗;患者膜性肾病病情稳定,监测血清Alb、SCr好转,且患者对肾脏穿刺并发症顾虑较大,遂未重复肾活检,建议定期监测肾功能、24 h尿蛋白。因iMCD难以解释反复肾周出血,需警惕肾盂周囊肿导致肾静脉压力升高继发出血可能,建议规律利尿、定期监测泌尿系影像学。出院随访至2021年10月29日,病情稳定。
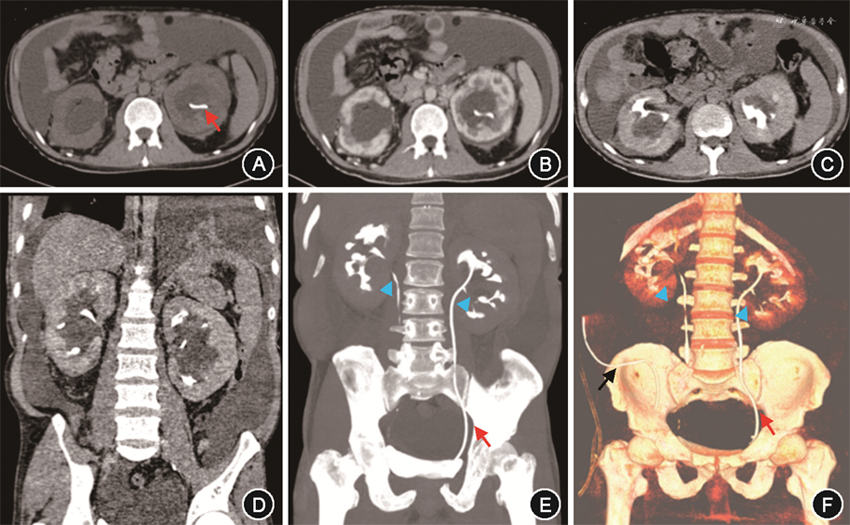
注:红色箭头示左侧D-J管;蓝色三角示肾盂受压狭窄处;黑色箭头示腹腔引流管
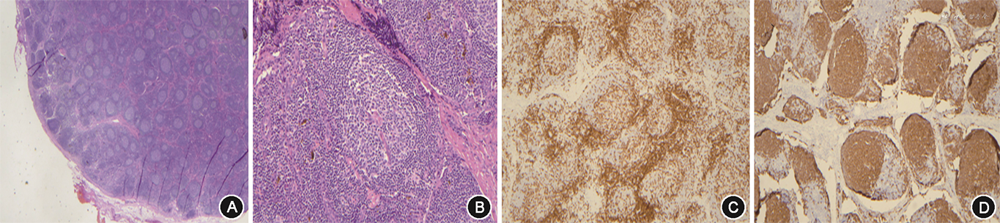
Castleman病临床表现复杂多变,需与恶性肿瘤、感染性疾病及自身免疫性疾病等鉴别,临床诊断困难,确诊依赖病理。
Castleman病造成肾盂周囊肿继发肾周出血系该病罕见表现,病程迁延复杂,患者辗转数年就诊国内多家医院,使临床诊断更为困难。
肾病综合征病因较多,需要全面分析。通过详细询问病史并排除感染、药物、自身免疫性疾病、肿瘤等继发因素后,才能诊断原发性肾病综合征。
临床中尽量用一元论解释病情。若一元论无法解释病情,需考虑二元论可能。患者同时存在两种罕见疾病概率较小,在诊断过程中需小心求证。
对于多系统受累、病程复杂迁延,病因不明的患者,多学科会诊有助于患者早期诊断,制定个体化治疗方案。
本例患者起病时有全身可凹性水肿、大量蛋白尿、低白蛋白血症等表现,符合肾病综合征诊断标准。继发肾病综合征常见病因包括:(1)系统性疾病:如糖尿病、系统性红斑狼疮、IgG4相关性疾病、血管炎性疾病等;(2)感染性疾病:包括梅毒、感染性心内膜炎、巨细胞病毒感染、乙型肝炎病毒感染、疟疾等;(3)药物:金制剂、锂制剂、汞、青霉胺、非甾体抗炎药等;(4)肿瘤:淋巴瘤、白血病、实体肿瘤、淀粉样变等。(5)遗传和代谢性疾病:如Alport综合征、Fabry综合征等。除外继发性因素后才可诊断原发肾病综合征。患者未充分筛查肾病综合征继发因素,仅根据肾脏穿刺病理予激素联合环磷酰胺治疗,病情缓解后未进一步规律随诊。
此后疾病逐渐进展,主要表现为多浆膜腔积液、反复肾周出血及多发淋巴结肿大,合并炎症因子升高、低白蛋白血症、肝功能不全、肾功能不全。患者此次入院24 h尿蛋白0.4 g,不符合肾病综合征诊断标准,但腹腔积液症状突出,以此作为诊断切入点。患者血清腹水白蛋白梯度(SAAG)>11 g/L。对于高SAAG型腹水常见病因包括心衰/缩窄性心包炎、下腔静脉梗阻、肝硬化、布加综合征、肝窦阻塞综合征等。但结合患者病史、临床表现及辅助检查,无上述常见病因相关提示。此外,还有约3% SAAG>11 g/L的患者可能由肿瘤、感染、自身免疫性疾病、腹腔脏器破裂(胃肠、胆囊等)、淋巴回流障碍(如Whipple病、丝虫病、结核等)、子宫内膜异位症等因素导致[1]。患者病程中无自身免疫性疾病、感染、腹腔脏器破裂、腹膜疾病等相关证据,病程整体迁延,而患者一般情况尚可,不符合常见恶性肿瘤临床特点;实验室检查方面提示多发肿大淋巴结、炎症因子升高,需警惕有无相对特殊血液系统疾病可能。遂行淋巴结活检,病理回报符合iMCD。
Castleman病是一种罕见血液系统疾病,又称为血管滤泡性淋巴结增生症。淋巴结病理检查是Castleman病诊断的金标准。根据淋巴结受累区域不同,可分为单中心型和多中心型Castleman病[2]。除淋巴结肿大外,多中心型Castleman病患者往往还伴有发热、盗汗、乏力、体重下降、皮疹、胸闷、憋气、贫血、高免疫球蛋白血症、肝功能不全、肾功能不全、容量负荷过多(全身水肿、胸水、腹水等)等临床表现[2, 3]。本例患者包括人疱疹病毒8型在内感染相关筛查均阴性,满足两条主要标准:(1)淋巴结病理符合Castleman病;(2)肿大淋巴结(短轴≥ 1 cm)≥2个淋巴结区域。满足4条次要标准:(1)ESR>15 mm/1h;(2)血清Alb<35 g/L;(3)估算肾小球滤过率<60 ml·min-1·(1.73m2)-1,24 h尿蛋白>150 mg;(4)浆膜腔积液。综上患者iMCD诊断明确[3]。部分iMCD患者临床表现具有“加重-缓解”趋势。根据国际Castleman病协作网络的危险度分层体系,患者为非重型iMCD。司妥昔单抗(IL-6单抗)是非重型iMCD一线治疗方案。一项随机双盲对照研究纳入79例iMCD患者,与安慰剂组相比司妥昔单抗组(11 mg/kg,每3周1次静脉输注)有34%的患者获得持续肿瘤及症状缓解。其他一线治疗方案包括沙利度胺-环磷酰胺-泼尼松(TCP方案)、司妥昔单抗/利妥昔单抗联合泼尼松治疗等[2]。若上述一线方案疗效欠佳或疾病进展,则可选择包括硼替佐米、西罗莫司、来那度胺等药物的单药或联合治疗。iMCD的核心治疗目标是控制高炎症状态。本例患者经积极对症治疗后,患者炎症状态明显好转,浆膜腔积液减少,肾功能恢复。综合实验室结果及临床表现,经血液科专科会诊考虑iMCD病情缓解,暂予继续对症利尿治疗,密切监测临床表现及实验室结果变化。
iMCD肾脏受累较为罕见,目前已有病例多为个案报道。肾脏受累患者常见临床表现为肾病综合征、慢性肾功能不全,也有患者表现为急性肾功能损伤、血尿、蛋白尿、水肿等[4],使用激素和(或)细胞毒性药物治疗后部分患者肾功能好转,临床症状改善,炎症因子(如CRP、IL-6、VEGF等)水平下降,但长期随诊肾脏受累整体表现为“缓解-加重”趋势[3]。综合患者病例特点及文献报道,考虑Castleman病继发肾病综合征及广泛炎症因子水平升高。目前已证实,IL-6调控异常及VEGF合成增多与Castleman病发病及继发肾脏受累相关[5, 6]。Castleman病肾脏受累,肾脏病理可表现为淀粉样变性、血栓性微血管病、微小病变肾病、膜性肾病、新月体肾炎、系膜增生性肾炎、局灶节段性肾小球硬化、膜增生性肾小球肾炎等[4,7, 8]。现已有Castleman病继发脑出血、蛛网膜下腔出血、腹膜后肿瘤出血的个案报道,推测可能与Castleman病导致血管病变(如血栓性微血管病、血栓、血管炎、血管壁薄弱)相关。但无论是哪种肾脏病理类型,目前未见肾周出血表现的报道。虽然Castleman病继发器官出血机制仍不明确,但依据目前临床资料尚不能完全除外患者反复肾周出血与Castleman病相关性。
自发性非创伤性肾脏包膜下和肾周间隙出血又称为Wunderlich综合征,是一种比较少见的临床情况,通常以急性腰痛起病,约20%患者可能进展为失血性休克[9]。肾脏肿瘤是Wunderlich综合征最常见病因,占整体病例的60%~65%,良性肿瘤以血管平滑肌瘤最常见,恶性肿瘤以肾细胞癌最常见。其他非肿瘤性病因包括:肾脏囊性病变、血管异常(如动脉瘤、肾脏动静脉畸形、血管炎、肾炎、肾静脉血栓等)、放射性损害、凝血功能障碍等。结合临床表现及辅助检查结果,考虑本例患者为肾盂周囊肿继发Wunderlich综合征可能。肾盂周围囊肿又称为肾盂周围淋巴管扩张或肾盂周围淋巴囊肿,其发病率为肾囊肿的4%~6%,常与肾盏相通或者囊肿内有间隔,因肾脏及肾周淋巴管回流至腹膜后淋巴管不畅所致,其壁为内皮细胞和淋巴细胞,囊腔内为淋巴液。影像学可表现为双肾体积增大、肾脏轮廓不规则,若囊肿进一步增大压迫肾门结构,存在肾静脉压力增高、肾静脉血栓、肾盂肾盏扩张、肾后性梗阻等风险[10]。目前肾盂周囊肿可选择经皮穿刺引流、囊肿硬化、囊肿剥离术、囊肿切除等方式治疗,若囊肿体积小、无临床症状,也可长期随诊观察[11]。治疗方面,肾盂周囊肿若合并肾周自限性出血,以补液、输血等保守支持治疗为主;若合并血流动力学不稳定,可考虑介入造影栓塞出血血管,必要时需行急诊肾部分/全部切除术。迄今仅有两例Castleman病继发淋巴管扩张报道,受累部位均为小肠,可能与肠系膜周及主动脉旁淋巴结肿大,继发淋巴回流障碍相关。对于本例患者,iMCD是否可能引起肾盂周囊肿,进而导致反复肾周出血还有待未来进一步研究。此外,在以往的诊治经验中,Casleman病造成腹腔积液多系VEGF增高引起,因此腹腔积液的生化特点更接近门脉高压依赖性腹腔积液,但也有部分患者随着病程发展出现了POEMS综合征[12]。这也提示,疾病是一个长期的动态发展的过程,临床随访十分重要,就像前辈大家张孝骞教授说的:一定要承认对诊断不能固定化,因为疾病并没有固定。随着疾病的发展和转化,诊断可以被证实、补充或推翻。这个认识不是一次完成的,它是反复的、动态的过程。患者动态地改变,医生的思想也随之改变[13]。
所有作者均声明不存在利益冲突

























