
噬血细胞综合征是一种由于各种诱因导致的细胞毒性T细胞和自然杀伤细胞过度活化,并刺激单核巨噬系统,分泌大量炎性细胞因子的危重疾病。淋巴瘤是导致继发噬血细胞综合征的重要病因之一。近年,随着对疾病认识的深入、检测手段的提高及新药的问世,淋巴瘤相关噬血细胞综合征的诊断和治疗体系逐渐完善。为更好地指导我国医师的临床实践,中国抗癌协会淋巴瘤专业委员会、中华医学会血液学分会淋巴细胞疾病学组以及中国噬血细胞综合征专家联盟,基于2018版《淋巴瘤相关噬血细胞综合征诊治中国专家共识》,结合新进展,对疾病的分类及治疗进行了修订,增加了中枢神经系统受累的解读,旨在规范和提高我国淋巴瘤相关噬血细胞综合征的诊治水平。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
噬血细胞综合征(hemophagocytic syndrome,HPS)又称噬血细胞性淋巴组织细胞增多症(hemophagocytic lymphohistiocytosis,HLH),是一种危重疾病,因各种诱因导致自然杀伤(natural killer,NK)细胞和细胞毒性T细胞(cytotoxic T lymphocytes,CTL)过度活化,并刺激巨噬细胞活化,分泌大量炎性细胞因子所致。淋巴瘤是继发HLH的常见诱因之一,发病率随着年龄的增长而增高[1]。随着对疾病认识的深入、检测手段的提高及新药的问世,淋巴瘤相关HLH的诊断和治疗不断改进和完善。本次专家共识主要对疾病的分类及治疗部分进行了修订,增加了HLH中枢神经系统受累的解读。专家共识的更新对于规范和提高我国淋巴瘤相关HLH(lymphoma associated HLH,LA-HLH)的诊治水平具有重要意义。
HLH是一组由原发或继发性免疫异常所致的过度炎症反应综合征。淋巴细胞、单核细胞和巨噬细胞系统异常激活、增殖,分泌大量炎性细胞因子,引起一系列炎症反应。临床以持续发热、全血细胞减少、肝脾肿大为主要表现,骨髓、淋巴结和肝脾组织中可见巨噬细胞吞噬血细胞现象(即噬血现象)。淋巴瘤诊疗过程中发生的HLH统称为LA-HLH。
LA-HLH可以根据发生的时间和诱因两种方法进行分类:根据发生时间的区别,可分为“淋巴瘤诱发的HLH”和“化疗期间的HLH”[3];根据HLH发生诱因的区别,可分为“淋巴瘤直接导致的HLH”“感染导致的HLH”及“免疫治疗诱发的HLH”。
1. 淋巴瘤直接导致的HLH:淋巴瘤疾病本身导致的HLH。HLH可发生于确诊淋巴瘤之前,也可与淋巴瘤诊断同时发生,或在淋巴瘤复发或进展时出现。淋巴瘤细胞分泌的细胞因子,如白细胞介素6(interleukin 6,IL-6)和干扰素γ(interferon-γ,IFN-γ)等导致的高炎症因子状态可能与HLH发生有关。这类HLH在T/NK细胞淋巴瘤最为多见,其次为弥漫大B细胞淋巴瘤和霍奇金淋巴瘤[4]。
2. 感染导致的HLH:多在淋巴瘤治疗过程中出现。患者在治疗后机体免疫功能受到抑制,常合并病毒感染、侵袭性真菌感染和一些细菌感染,在感染的刺激下出现HLH的临床表现。该类型特别要关注EB病毒(Epstein-Barr virus,EBV)感染。
3. 免疫治疗诱发的HLH [5, 6, 7, 8]:淋巴瘤治疗过程中出现,多发生于嵌合抗原受体(chimeric antigen receptor,CAR)T细胞(CAR-T)疗法或免疫检查点抑制剂(immune checkpoint inhibitors,ICIs)治疗后,亦可见于免疫调节剂、双特异性抗体(bispecific T-cell engagers,BiTE)等治疗后。欧洲血液和骨髓移植学会(European Society for Blood and Marrow Transplantation,EBMT)成员中心回顾性资料显示,CAR-T治疗后HLH发生率为3.48%,其表现与巨噬细胞活化综合征(macrophage activation syndrome,MAS)/HLH类似。ICIs应用后HLH发生可能与治疗导致T细胞异常活化有关,世界卫生组织(World Health Organization,WHO)个人病例安全报告(individual case safety reports,ICSRs)数据显示,ICIs应用后HLH发生率存在地区差异,为0.03%~0.40%,含细胞毒T细胞相关抗原4(cytotoxic T lymphocyte antigen-4,CTLA-4)方案发生率高于程序性死亡受体1(programmed death receptor-1,PD-1)/程序性死亡受体配体1(programmed death receptor ligand-1,PD-L1)抑制剂;HLH占血液系统不良反应的15%,与其他血液学毒性相比,发病早且死亡率高。
(一)LA-HLH诊断标准:推荐采用国际组织细胞协会于2004年修订的HLH-2004标准[9]。在具有明确病理诊断的淋巴瘤的基础上,除外原发HLH,符合HLH-2004诊断标准8条指标中的5条或以上,LA-HLH的诊断可以成立。
HLH-2004诊断标准具体如下:符合以下8条指标中的5条或以上:(1)发热:体温>38.5 ℃,持续>7 d;(2)脾大;(3)血细胞减少(累及外周血两系或三系):血红蛋白<90 g/L(<4周龄婴儿,血红蛋白<100 g/L),血小板<100×109/L,中性粒细胞<1.0×109/L且非骨髓造血功能减低所致;(4)高甘油三酯血症和(或)低纤维蛋白原血症:甘油三酯>3 mmol/L或高于同年龄的3个标准差,纤维蛋白原<1.5 g/L或低于同年龄的3个标准差;(5)在骨髓、脾、肝脏或淋巴结中发现噬血现象;(6)NK细胞活性降低或缺如;(7)血清铁蛋白升高:铁蛋白≥500 μg/L;(8)可溶性白细胞介素-2受体(soluble CD25,sCD25)升高。
1. 虽然有学者提出了HLH评分系统HScore及针对LA-HLH亚型的诊断标准[11, 12],但尚未得到广泛公认。HLH-2004诊断标准仍是目前临床诊断HLH应该遵循的原则。
2. 淋巴瘤疾病本身与HLH在临床特征上有很多交叉重叠之处[如发热、血细胞减少、肝脾肿大、铁蛋白升高、乳酸脱氢酶(lactate dehydrogenase,LDH)升高等],对于临床进展快、短期内出现器官功能衰竭的患者,需警惕HLH可能。但应避免诊断扩大化,各项指标(如sCD25、血清铁蛋白和LDH等)的动态监测更为重要。在淋巴瘤患者组织中找到噬血现象,高度提示HLH的可能,sCD25/血清铁蛋白比值显著升高也是诊断LA-HLH的预警指标[13]。HLH相关细胞因子谱,如IL-6、IL-10等[14],可以协助鉴别淋巴瘤患者是否同时合并了HLH。
3. 正电子发射计算机断层显像(positron emission tomography-computed tomography,PET/CT)不能用于LA-HLH及其他类型HLH的鉴别诊断,但可协助指导选取活检部位[15]。流式细胞术(flow cytometer,FCM)可以快速发现骨髓中异常表型淋巴细胞,提示存在淋巴瘤可能[16],但病理诊断仍是确诊金标准。
4. CAR-T治疗相关HLH的诊断尚缺乏统一标准,多数仍沿用HLH-2004标准。目前也有学者应用2018年MD Anderson癌症中心提出的针对CAR-T治疗相关HLH的诊断标准[6],即细胞因子释放综合征(cytokine release syndrome,CRS)过程中铁蛋白升高>10 000 μg/L,伴以下至少两种器官毒性[常见不良反应事件评价标准(common terminology criteria for adverse events,CTCAE)4.0]:(1)≥3级血清胆红素、天冬氨酸转氨酶或丙氨酸转氨酶水平升高;(2)≥3级少尿或血清肌酐水平升高;(3)≥3级肺水肿;(4)骨髓或器官中存在噬血细胞。ICIs相关HLH仍沿用HLH-2004诊断标准,国内专家对于其诊治亦提出了指导建议[17]。
当患者出现意识障碍、头痛、头晕、癫痫发作、易激惹、精神异常、肢体活动异常等症状时,需考虑CNS受累可能。应积极完善脑脊液检查及头颅影像学检查,区分患者为淋巴瘤CNS受累、CNS感染、HLH-CNS受累或CAR-T治疗相关脑病综合征(CAR T-cell related encephalopathy syndrome,CRES)。脑脊液检查建议完善脑脊液常规、生物化学、细胞学(淋巴瘤细胞及噬血细胞)和病原学检查,脑脊液细胞因子、铁蛋白及sCD25检查可协助进一步鉴别是否存在HLH-CNS受累。脑脊液FCM可协助快速发现异常表型淋巴细胞。影像学检查首先推荐磁共振成像(magnetic resonance imaging,MRI)。
HLH疾病进展迅速,死亡率高,因此及时发现HLH疑似患者,给予正确诊断至关重要。淋巴瘤与HLH既可以同时出现,也可能先后出现,两者之间既相互独立又密切相关。因此如何从淋巴瘤患者中发现HLH,以及从HLH患者中寻找潜在淋巴瘤是临床工作的重点。诊断LA-HLH建议遵循以下流程。
1. 及时发现疑似HLH患者并评估:淋巴瘤患者病程中出现持续发热、血细胞减少、肝脾肿大或不明原因的肝衰竭等HLH疑似症状,可按以下流程明确是否合并HLH(图1)。首次评估未满足诊断标准的疑似患者,需动态监测各项指标,短期内再评估。
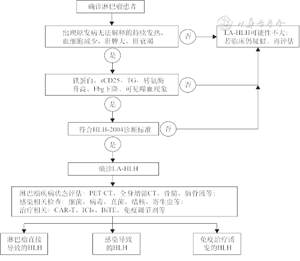
注:sCD25为可溶性白细胞介素-2受体;TG为甘油三酯;Fbg为纤维蛋白原;PET/CT为正电子发射计算机断层显像;CAR-T为嵌合抗原受体T细胞;ICIs为免疫检查点抑制剂;BiTE为双特异性抗体
2. 判断HLH的诱因:确诊LA-HLH后需完善HLH诱因相关检查(图1),指导后续治疗。诱因可能单独存在,也可能二者或三者同时存在,需进行全面评估。
详见《中国噬血细胞综合征诊断与治疗指南(2022年版)》[23],补充说明如下[3,24, 25]。
1. 关于EBV:EBV感染参与在各种类型HLH的疾病过程中,并非仅见于EBV-HLH。在LA-HLH患者中,其可能是HLH的感染诱因,亦可能是EBV阳性淋巴瘤的疾病表现。对于外周血EBV-DNA阳性患者,建议完善EBV感染淋巴细胞亚群检测,协助判断为治疗后机体免疫功能受抑出现的EBV再激活,还是淋巴瘤进展所致。
2. 关于基因缺陷:HLH患者确诊淋巴瘤后,若HLH相关功能学检查存在异常、有家族史或HLH反复发作时,推荐进行基因评估。HLH相关功能学检查建议包括NK细胞活性和脱颗粒功能检测(NK细胞和CTL细胞膜ΔCD107a),颗粒酶B、穿孔素、UNC13D、SAP、XIAP等蛋白表达量的检测。(1)许多HLH相关基因缺陷增加了淋巴瘤发生的风险[26, 27],如SH2D1A、XIAP、MAGT1、CD27、CD70等;当HLH患者同时存在已知的HLH缺陷基因和淋巴瘤时,仍应诊断为原发性HLH。(2)部分淋巴瘤患者存在的遗传学异常可能与HLH高发相关[28, 29],如ECSIT-T419C突变与结外鼻型NK/T淋巴瘤发生HLH有关;存在TIM-3胚系突变的皮下脂膜炎样T细胞淋巴瘤患者较TIM-3无突变的患者更易发生HLH。
LA-HLH的治疗分为两个方面[3,22]:一方面是针对HLH的治疗,控制炎症反应,改善器官功能障碍,控制HLH活化进展;另一方面是针对淋巴瘤的治疗,消除HLH诱因,防止HLH复发。
1. 对于LA-HLH的治疗应该先针对 HLH还是先针对淋巴瘤,目前尚无循证学依据,需根据患者的不同状况决定。本专家共识推荐给予HLH诱因指导下的分层治疗,具体可参考图2。
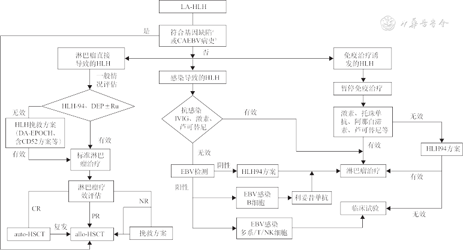
注:a符合原发HLH诊断;b确诊淋巴瘤前的既往病史;CAEBV为慢性活动性EB病毒感染;DEP为脂质体阿霉素+依托泊苷+甲泼尼龙方案;Ru为芦可替尼;DA-EPOCH为剂量调整EPOCH方案;CD52单抗为阿伦单抗;CR为完全缓解;PR为部分缓解;NR为无治疗反应;auto-HSCT为自体造血干细胞移植;allo-HSCT为异基因造血干细胞移植;IVIG静脉输注丙种球蛋白;EBV为EB病毒
2. 补充说明:(1)对于“淋巴瘤直接导致的HLH”,需评估患者器官功能;对于器官功能尚可的患者,推荐给予兼顾HLH及淋巴瘤的含依托泊苷的联合化疗方案,如DEP(脂质体阿霉素、依托泊苷、甲泼尼龙)、DA-EPOCH(依托泊苷、阿霉素、长春新碱、环磷酰胺、泼尼松)或DEP样方案;对于器官功能较差的“脆弱”患者,可考虑给予HLH-94方案或非细胞毒性药物治疗。HLH得到控制后应积极过渡到标准的淋巴瘤化疗,具体参见《中国淋巴瘤治疗指南》[30]。
(2)对于“感染导致的HLH”和“免疫治疗诱发的HLH”,HLH缓解、感染等诱因祛除后,可回归至既往的淋巴瘤治疗。
(3)LA-HLH诊治过程中,上述诱因可能同时或者先后出现,对于HLH治疗效果欠佳或者复发的患者,需再次进行诱因评估指导治疗,并判断患者是否需早期行异基因造血干细胞移植(allogeneic hematopoietic stem cell transplantation,allo-HSCT)。
1. HLH-94方案:HLH-94及HLH-2004方案是目前广泛应用的标准HLH治疗方案,由国际组织细胞协会于1994年制定[31],2004年修订[9]。根据HLH-94和HLH-2004治疗方案的前瞻性临床研究结果,国际组织细胞协会不推荐LA-HLH患者使用HLH-2004方案进行诱导治疗。HLH-94方案详见《中国噬血细胞综合征诊断与治疗指南(2022年版)》[23]。对于成人、尤其是伴有合并症的老年人[32],2019年国际组织细胞协会成人HLH专家共识[33]提出依托泊苷(etoposide,VP-16)用药频率可以减至每周1次,剂量可以从150 mg/m2减至50~100 mg/m2。
2. DEP方案:DEP方案是由脂质体多柔比星、VP-16和甲泼尼龙组成的三药联合化疗方案[34]。LA-HLH患者推荐给予剂量调整DEP方案(NCT04077905):脂质体多柔比星35 mg·m-2·d-1,第1天。VP-16 100 mg·m-2·d-1,第1天(可根据患者器官功能及年龄调整剂量)。甲泼尼龙2 mg·kg-1·d-1,第1~3天;0.75 mg·kg-1·d-1,第4~7天;0.25 mg·kg-1·d-1,第8~10天;0.1 mg·kg-1·d-1,维持至下一疗程;该方案可2周重复一次。与芦可替尼(ruxolitinib,Ru)或左旋门冬酰胺酶(L-asparaginase,L-asp)联合应用时,脂质体多柔比星可减量至25 mg/m2。DEP方案可用于LA-HLH的初始诱导治疗,也可用于HLH-94方案治疗无效的难治性患者。研究证实,DEP方案初始诱导治疗的2周及4周总治疗反应率(overall response rate,ORR)均优于HLH-94方案;在HLH-94方案无应答的难治性LA-HLH患者中也有较好的二次应答率[35, 36]。
3. 其他含依托泊苷的化疗方案:对于体能状态较好的“淋巴瘤直接导致的HLH”,含VP-16的多药联合化疗方案[37, 38],如Ru-DEP、L-DEP(L-asp联合DEP)、Ru-DED(脂质体阿霉素、依托泊苷、地塞米松)、DA-EPOCH,疗效可能优于HLH-94方案。与DEP方案联合应用时,芦可替尼的成人推荐剂量为10 mg/次、2次/d。
4. 其他:HLH的治疗新药物,如Janus激酶(JAK)1/2抑制剂(芦可替尼)、IFN-γ抑制剂(伊帕伐单抗)、CD52单抗(阿伦单抗)、IL-1受体拮抗剂(阿那白滞素)等均在HLH患者中获得了一定的疗效[39, 40, 41, 42],但在LA-HLH中尚缺乏前瞻性临床研究。细胞因子吸附治疗(细胞因子吸附柱或血浆置换)可能短期改善患者症状及器官功能[43]。
5. 中枢神经系统治疗:中枢神经系统累及患者,给予腰椎穿刺及鞘内注射,推荐选择含可透过血脑屏障的药物的方案治疗。
1. 明确诊断为原发HLH的淋巴瘤患者,以及有明确慢性活动性EBV感染(chronic active EBV infection,CAEBV)病史的淋巴瘤患者,推荐早期行allo-HSCT。移植应在患者HLH达到临床缓解后及早进行,发病至移植的时间是影响预后的一个重要因素[44]。HLH反复发作,合并HLH的难治/复发淋巴瘤、高度侵袭性淋巴瘤(NK/T淋巴瘤、伯基特淋巴瘤)可考虑进行allo-HSCT[45, 46]。
2. 对于“淋巴瘤直接导致的HLH”患者,经标准淋巴瘤治疗后,进行淋巴瘤疗效评估,完全缓解(complete remission,CR)患者,若能够耐受强化治疗,推荐行自体HSCT(autologous HSCT,auto-HSCT)。部分缓解(partial response,PR)及无反应/疾病进展(no response/progressive disease,NR/PD)患者可考虑进行allo-HSCT,在移植前可给予挽救治疗方案降低肿瘤负荷。对于“感染导致的HLH”和“免疫治疗诱发的HLH”,HLH缓解后,淋巴瘤移植推荐参考《中国淋巴瘤治疗指南》[30]。
3. allo-HSCT预处理方案及供者选择:虽然在原发HLH及非恶性疾病相关HLH患者中减低强度预处理(reduced-intensity conditioning,RIC)似乎优于清髓预处理(myeloablative conditioning,MAC),但对于LA-HLH,仍推荐根据淋巴瘤疾病特点选择预处理方案,MAC可能更有利于控制原发病[33]。在选择亲缘供者时应全面评估供者的NK细胞活性和脱颗粒功能,进行HLH缺陷基因蛋白表达水平检测及HLH缺陷基因筛查,并检测外周血单个核细胞及血浆/血清EBV-DNA。
HLH患者常合并感染及多脏器功能受累。支持治疗的原则与allo-HSCT患者相似,需包括真菌、病毒及卡氏肺子虫感染的预防,防范中性粒细胞减少,必要时给予静脉注射丙种球蛋白(intravenous immunogloblin,IVIG)。发生新出现的发热时,需考虑机会性感染及HLH复发的可能,并给予经验性广谱抗生素治疗[3]。
HLH患者常存在严重的血小板减少和凝血功能异常,自发性出血风险高。治疗期间建议将血小板计数维持在30×109/L以上。对于急性出血患者可输注血小板、新鲜冰冻血浆、凝血酶原复合物、纤维蛋白原等,必要时补充活化Ⅶ因子。粒系集落刺激因子需谨慎使用,避免应用粒细胞巨噬细胞集落刺激因子。重组人血小板生成素、艾曲波帕、海曲波帕等可在HLH治疗期间用于提升血小板计数。
LA-HLH患者可能在疾病过程中,由于炎症反应或可能的药物毒性损害,出现或发展为心肺、肝、肾等多脏器功能不全。治疗前应充分评估患者的脏器储备功能,并给予适当的对症支持治疗,治疗过程中严密监测脏器功能。
HLH诱导治疗期间,建议每2周进行一次疗效评估,疗效评价标准见表1[47]。淋巴瘤疗效评估参考《中国淋巴瘤治疗指南》[30]。

噬血细胞综合征(HLH)疗效评价标准[47]
噬血细胞综合征(HLH)疗效评价标准[47]
| 疗效 | 标准 |
|---|---|
| CR | sCD25、血清铁蛋白、血细胞计数、甘油三酯、噬血现象、意识水平(有CNS-HLH者)均恢复正常范围 |
| PR | sCD25、血清铁蛋白、血细胞计数、甘油三酯、噬血现象、意识水平(有CNS-HLH者)中≥2项症状/实验室指标改善25%以上, 个别指标需达到以下标准: (1)sCD25水平下降1/3以上; (2)铁蛋白和甘油三酯下降25%以上; (3)不输血情况下:中性粒细胞<0.5×109/L者需增加100%并>0.5×109/L,中性粒细胞0.5~2.0×109/L者需增加100%并恢复正常; (4)丙氨酸转氨酶>400 U/L者,需降低50%以上 |
| NR | 未达到上述标准 |
注:CR为完全缓解;PR为部分缓解;NR为无治疗反应;sCD25为可溶性白细胞介素-2受体;CNS-HLH为中枢神经系统HLH
未经及时诊断及治疗的LA-HLH,中位生存时间低于2个月[22]。与同类型淋巴瘤患者相比,合并HLH的患者预后更差,生存时间更短。早期诊断、分层治疗,给予积极的器官功能支持,可为患者后续治疗创造条件。HSCT能够改善部分患者预后。
共识编写委员会名单:
执笔专家:王昭(首都医科大学附属北京友谊医院血液科);石远凯(中国医学科学院肿瘤医院肿瘤内科)
参与讨论专家(按照姓氏拼音排序):白鸥(吉林大学第一医院肿瘤中心血液科);郭涛(华中科技大学同济医学院附属协和医院血液科);黄慧强(中山大学附属肿瘤医院肿瘤内科);黄文荣(解放军总医院第五医学中心血液科);江明(新疆医科大学附属第一医院血液科);江倩(北京大学人民医院血液病研究所);克晓燕(北京大学第三医院血液科);李菲(南昌大学第一附属医院血液科);李建勇(南京医科大学第一附属医院血液科);刘丽宏(河北医科大学第四医院血液科);刘利(空军军医大学第二附属医院血液科);牛挺(四川大学华西医院血液科);任汉云(北京大学第一医院血液科);沈建箴(福建医科大学附属协和医院血液科);石远凯(中国医学科学院肿瘤医院肿瘤内科);宋玉琴(北京大学肿瘤医院淋巴肿瘤内科);孙秀华(大连医科大学附属第二医院肿瘤内科);谭获(广州医学院第一附属医院血液科);王昭(首都医科大学附属北京友谊医院血液科);魏娜(首都医科大学附属北京友谊医院血液科);徐兵(厦门大学附属第一医院血液科);薛宏伟(青岛大学附属医院淋巴瘤科);张会来(天津医科大学肿瘤医院淋巴瘤科);张明智(郑州大学第一附属医院肿瘤科);张清媛(哈尔滨医科大学附属肿瘤医院肿瘤内科);张曦(陆军军医大学第二附属医院血液科);赵东陆(哈尔滨血液病肿瘤研究所);赵维莅(上海交通大学医学院附属瑞金医院血液科);曾云(昆明医科大学附属第一医院血液科);周凡(解放军北部战区总医院血液科);周辉(湖南省肿瘤医院淋巴瘤血液内科);周剑峰(华中科技大学同济医学院同济医院血液科);朱尊民(河南省人民医院血液科)
所有作者均声明不存在利益冲突

























