
宏基因组测序技术在未知病原体感染以及危重症感染诊断中的临床应用价值日益凸显。由于宏基因组测序庞大的数据量以及临床诊疗的复杂性,使得宏基因组测序在实际应用中存在着数据分析困难、不易解读的难题。因此,在临床实践的过程中,把握生物信息学分析的关键点、建立标准化的生物信息学分析流程至关重要,这是宏基因组测序由实验室走向临床转化的重要步骤。目前,宏基因组测序的生物信息学分析已经有了长足的进步,但随着临床对生物信息学分析规范化、标准化的高要求以及计算机技术的发展,宏基因组测序的生物信息学分析也面临着新的挑战。本文主要从质量控制、致病菌的判定以及可视化等方面进行阐述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
宏基因组高通量测序(metagenomics next generation sequencing,mNGS)以生物样本中的核酸为检测对象,通过核酸片段的物种代表性来鉴别生物样本中的物种组成。作为一种非靶向核酸检测方法,mNGS理论上可无偏倚地获得样本中所有物种的基因组信息。mNGS在检测感染性疾病致病病原体临床应用中受到广泛重视。以新型冠状病毒的发现过程为例,mNGS在针对新发、突发、复杂及混合感染相关病原体的实验室诊断中,发挥着越来越重要的作用,其临床参考价值也更为突出。但在临床应用中不得不正视mNGS的生物信息学分析相较于其他高通量测序相关分析方法具有更高的复杂性。原因包括:(1)数据复杂性高:送检临床样本中的微生物种类繁多,且不同物种的基因组信息没有固定的组成比例,加之不同类型样本携带的复杂人源信息,直接导致了mNGS所获得的基因组信息极其复杂;(2)分析维度多:在临床实际应用中,mNGS除了具有不同级别微生物物种鉴别的常规功能之外,还涉及耐药/毒力基因的分析等任务;(3)分析速度要求快:在面对各种疑似感染病症,尤其是急危重症患者时,临床始终有快速、准确鉴定病原体的迫切需求。因此,如何利用生物信息学分析这把利剑,在海量的测序数据中快速、准确地获取与临床感染直接相关的若干种病原微生物成为了mNGS走向临床应用的基础。不仅如此,伴随着计算机技术的快速发展以及临床对更多诊疗信息的渴求,对mNGS数据的生物信息分析流程提出了更高的要求,同时也是对生物信息分析结果的临床解读提出了新的挑战。
生物信息学分析是宏基因组测序应用的重要组成部分,是对测序产出的原始数据进行处理和分析的过程。在mNGS应用于临床病原学诊断前,研究人员已开发了多款宏基因组生物信息分析算法及软件应用于科学研究[1]。这些算法主要是针对微生物种群多样性、物种组成、基因相互作用和功能预测进行设计的,并且是基于多样本的统计分析。与科研用途的生物信息分析流程不同,临床用途的mNGS生物信息分析侧重于关注感染患者的样本中可能存在病原体的检测信息,更强调流程标准化、样本间分析的独立性。
目前国内尚无国家药品监督管理局批准的临床mNGS生物信息学分析的标准分析流程,临床mNGS生物信息学分析的流程普遍是实验室自建方法。针对mNGS进行的数据分析,不同实验室开发的算法不同,其流程模块也不尽相同,因此建立模块化的标准生物信息分析流程,并进行高质量的分析性能验证对于mNGS的临床应用管理至关重要[2]。临床mNGS生物信息学分析流程通常由数据预处理、宿主基因组数据过滤、微生物序列识别及物种鉴定、阳性病原体判定四大模块组成。
通过临床mNGS生物信息学分析流程,复杂的测序数据得以转化为以微生物为对象的数据,并实现指标数字化。1份合格的mNGS分析报告离不开生物信息分析流程中各个环节的质量评估和审核,因此把握生物信息学分析的关键点是mNGS能够提供准确结果、服务好临床的重中之重。
1.测序数据质控参数:高质量的测序数据是生物信息学分析产出准确结果的基础。测序数据质量控制的目的是保证下机数据符合湿实验的测序策略,保证数据质量满足后续的生物信息学分析。临床测序数据质量控制的重点是建立符合临床实际的质量控制体系,关键是确定合理的质量评价指标。评价测序数据质量的主要指标包括:样品测序数据量、碱基质量、有效测序长度、测序标签跃迁率等。目前已有多款不同的生物信息开源软件可用于测序数据评价,如FastQC、ngsReports、MultiQC等,以及低质量序列修剪或去除软件,如fastp、PRINSEQ、Trimmomatic等。
根据文献报道[3],临床mNGS数据的基本质控指标应达到:碱基质量(Q30)≥80%、有效序列长度≥50 bp;控制测序标签跃迁导致的数据污染;测序数据量需根据不同样品类型评估,目前尚无统一标准。
2.数据库与算法:mNGS生物信息数据分析和处理的原理是通过序列比对算法将测序数据与已知物种的参考基因组数据库进行同源性分析,达到鉴定微生物物种的目的。由此可见,数据库和算法的选择对生物信息分析流程的性能和结果准确性有很大的影响。
数据库可分为宿主基因组数据库和微生物检测数据库两类,分析流程应使用公认的或经过文献验证的公共数据库;有条件可在公共数据库的基础上,根据临床实践搭建二级数据库。由于人类基因组序列常包含高多态性区域、高复杂度区域、微生物高同源性区域等,高度的复杂性易造成宿主基因组过滤不充分或者过度过滤的情况,从而导致微生物基因组分析结果存在假阳性和假阴性[4, 5]。因此,宿主基因组数据库的构建、特殊区域或序列的标记尤为重要[6]。宿主的参考基因组来源常选用参考基因组联盟人类基因组38(GRCh38,https://www.ncbi.nlm.nih.gov/grc/human/data?asm=GRCh38.p14)、参考基因组联盟人类基因组37(GRCh37,https://www.ncbi.nlm.nih.gov/grc/human/data?asm=GRCh37.p13)、人线粒体参考基因组(HmtD,https://www.hmtdb.uniba.it),根据临床需要,可补充选择中国人标准基因组(CNPhis0000542,https://db.cngb.org/search/project/CNPhis0000542)、端粒到端粒基因组(CHM13,https://github.com/marbl/CHM13)等。
目前,微生物检测数据库来源主要有美国国家生物信息中心(National Center for Biotechnology Information,NCBI)、美国病原体系统资源整合中心(Pathosystems Resource Integration Center,PATRIC)、真核生物病原体数据库(Eukaryotic Pathogen Database,EuPathDB)、病毒病原数据分析资源库(Virus Pathogen Resource,ViPR)等。但公共数据库的微生物基因组质量良莠不齐,存在物种分类错误、测序错误、非目标物种的序列污染等问题。另一方面,临床病原微生物存在季节和地域性偏差,进化速度较快,可能与公共数据库收录的微生物参考基因组存在差异。因此,公共数据库一般要进行基因组或序列层面筛选、甄别、整合,以构建专用于临床鉴定的二级微生物检测数据库。
微生物物种鉴定算法是mNGS生物信息分析流程中的核心模块,根据测序范围不同,物种鉴定方法可以分为标记基因分类和完整基因组分类两种,这两种方法最大的区别是所采用的分析算法不同,详见表1。建议按照研究目标和资源选择适当的分析算法。对于检测目标为样本中优势菌群的研究,采用标记基因序列分类方法就可以满足要求。在急危重症患者的感染源检测中,对物种鉴定精度、时效性要求较高,则需采用经典比较算法,同时采用中央处理器(central processing unit,CPU)和内存,利用分布式计算、云计算和利用图形处理器(graphics processing unit,GPU)或现场可编程逻辑门阵列(field programmable gate array,FPGA)加速等方法进行加速,以达到临床需求。总之,从临床mNGS的检测需求出发需要达到以下两个目标:第一,需要实现在微生物种水平,特殊情况下甚至需要在亚种水平对样本中的微生物进行鉴定;第二,需要有可以体现半定量、相对定量参考的指标,以提示鉴定微生物在样本中的优势程度。
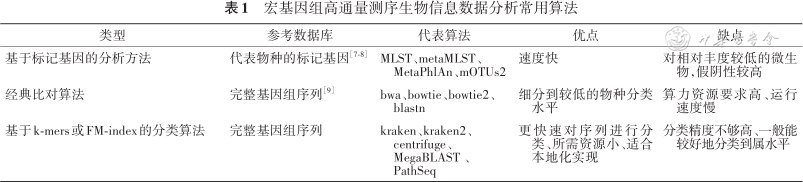
宏基因组高通量测序生物信息数据分析常用算法
3.致病菌判定:致病菌的判定是临床mNGS分析的重点,也是生物信息学分析的难点,单纯以微生物序列数排前的分析方法并不能有效找出真正致病菌[10]。生物信息学报告可以提供多维度的指标来判定微生物,包括按单一物种的序列数、特异序列数、覆盖长度、平均覆盖深度、覆盖均一度等;按样品类型计算物种的相对丰度,判断疑似定植菌;以及按批次和样品类型计算疑似污染序列(例如强阳性)和疑似环境背景菌等。但由于人体微生态的多样性以及感染诊断的复杂性,往往不能仅从mNGS分析结果中判定致病菌,而是需要在mNGS病原学提示的基础上,结合临床症候、感染指标、影像学特征进行报告解读。因此,建议具有临床微生物知识和感染诊断经验的医师共同参与其中,结合生物信息学分析、其他实验室相关指标和临床症状、体征,才能准确判断致病菌,进而针对病因开展治疗。临床mNGS检测报告中还包含微生物耐药或毒力基因等辅助信息,目前对这些数据的使用还处于探索阶段,使用过程中要保持较为谨慎的态度[11]。
4.可视化:生物信息学是一个复杂且专业性较高的学科,现有的生物信息学分析对于常规临床诊疗行为来说仍存在一定的技术壁垒。交互式、可视化的生物信息分析软件可显著降低mNGS临床应用的门槛。可视化分析包括流程可视化和数据可视化两个维度。流程可视化指可以在界面上配置、监控模块化的生物信息分析流程,提高mNGS临床应用中的标准化程度。数据可视化则能够将抽象的数据自动转化为有意义的分析图表,使分析结果更易懂、易用。在mNGS的检测、分析过程中,可以将需要进行可视化的数据分为维度数据、层次数据和网络数据等类型;维度数据又包括一维数据、二维数据、三维数据和多维数据。
目前,多维数据、层次数据和网络数据是可视化研究的热点。最经典的多维数据可视化方法是平行坐标,其优点为原理简单、便于操作。但在处理临床多维数据时,由于折线过于密集,就会引起“视觉混淆”,需要通过聚类、维度重排、拓朴等方法进行改进,例如mNGS临床研究中对微生物组成的主成分分析(principal component analysis,PCA)。此外,还可以用图表和动画等形式展示多维数据。层次数据具有明确的等级或层级关系,比如生物界门纲目科属种的划分,常使用节点链接图和树图进行数据可视化。网络数据是指数据间形成关系网络的数据集,在宏基因组研究中较为常见的为同一样本中微生物与微生物之间的共生/抑制关系网络。这类数据呈现复杂的网状结构,需要使用自动布局算法来实现数据可视化,较为经典的算法包括力导向布局、分层布局和网格布局。
自适应可视化是生物信息可视化的重要发展方向。在测序流程和数据标准化基础之上,生物信息分析软件可以根据数据特征和临床需求自动生成展示方案。
现阶段,mNGS已经逐渐走入临床应用,成为感染性疾病病原学诊断的重要方法之一。但在生物信息分析技术的临床应用过程中,仍面临着诸多挑战和创新。
1.生物信息分析的质量控制:目前国内外缺乏针对mNGS生物信息分析方法的质量控制和评价体系。2019年开始,国家卫生健康委临床实验中心开展组织了多次宏基因组病原检测的室间质评测试,从整体角度对各种mNGS方法的检测性能进行了评测,结果显示不同单位的报告性能差异很大[12, 13],表明建立生物信息分析流程的系统性评测体系和标准迫在眉睫。加强流行病学中观察性研究报告质量指南(Strengthening the Reporting of Observational Studies in Epidemiology,STROBE)和美国病理协会等学术组织也提出了针对mNGS生物信息分析流程验证的建议[14],主要目的是:(1)验证进化上近源物种的识别能力,特别是在来自某个物种的序列数很高的情形下;(2)确认检出灵敏度,即病原体序列数的检出下限(limit of detection,LOD);(3)验证数据库的完整性,确认重要的病原体是否都能被准确地检出。
在实际工作中,建立验证mNGS生物信息分析方法性能的模拟样本是mNGS生物信息分析质量控制的重要步骤[15]。目前模拟样本的构建主要是基于已知常见病原体基因组数据和人基因组数据,模拟二代测序原理,按不同样本中人源基因组和微生物基因组的理论比例混合构建数据集。该方法构建的模拟样本可实现对常见样本、常见病原体的生物信息分析性能验证。但是这种方法仍不可替代真实样本,需要尽快构建基于真实样本的参考数据,进而构建算法质量控制的基础。建议在未来的质量控制中细化不同环节的性能和质量评测,如增设相同的测序模拟样本用以评测不同生物信息分析流程的性能。
2.宿主信息的挖掘:宏基因组测序数据包括样本中宿主和微生物的信息。当前生物信息分析流程主要集中于微生物信息的分类和鉴别,但近年来很多研究尝试挖掘宏基因组测序宿主信息中有临床价值的信息,提示未来的生物信息分析可从宿主信息挖掘方面进行研发,从而为临床决策提供更全面的参考信息。
病原感染是宿主免疫系统与病原微生物之间的复杂交互过程,宿主信息在病原感染的判断中可以发挥重要功能。2020年,Mayhew等[16]提出由29个mRNA分子组成的分类器,用于区分细菌和病毒感染。2021年,Zhang等[17]的研究采用宏基因组和宏转录组测序分析,发现新型冠状病毒感染患者的宿主免疫反应通路产生明显变化。如何区分检出微生物是定植菌还是感染相关的病原体,是mNGS的一大挑战。2018年,Langelier等[18]开发了基于规则和逻辑回归的两套模型,通过结合宿主反应和病原检出信息,用于区分潜在病原和呼吸道定植菌,可达到95%的准确率。
除此之外,mNGS本身作为不明原因发热等疑难病症在感染方面的重要病因筛查工具,海量的患者基因组信息可以同步进行染色体不稳定性分析,能够提供肿瘤信号预警信息[19]。2021年,Guo等[20]发现来自mNGS的宿主序列可以用于检测肿瘤患者的染色体不稳定性,从而可以有效地对影像学异常,如肺部占位性病变的患者提供辅助诊断。因此充分利用生物信息学全面分析mNGS的微生物数据和宿主信息,能够为患者提供更多、更全面的诊疗信息。
3.人工智能:人工智能技术的发展为基于组学数据的临床检验提供了创新性的思路。Liu等[12]通过机器学习算法,采用假阳性滤过器,有效降低了mNGS结果中的假阳性物种。Xu等[21]利用深度学习算法,从宏基因组数据中高效识别多种微生物抗菌肽,助力下一代抗菌剂研发。海量复杂的mNGS数据正是人工智能施展本领的绝佳领域。通过挖掘信息的潜在关联,构建逻辑认知,可以将所有与病原学诊断相关的信息连接起来,构建关系网络,进而从“关系”的角度多维度分析问题。但是人工智能在mNGS中的应用研究尚处于初始阶段,这就需要在实践中加强生物信息学分析与临床信息、宏基因组测序与大数据分析等交叉学科的人才培养,运用跨专业科研团队的力量迎接这一挑战。
综上所述,随着mNGS技术在临床应用的普及,生物信息学分析将成为该技术实现临床落地的重要一环。建立标准化的生物信息分析流程和从临床出发的性能评估方法是mNGS走好临床应用之路的关键;借助人工智能等新技术整合多维度的信息,将感染诊断带入数字化时代,依托生物信息分析实现更精准、更快捷的mNGS结果解读是助力临床感染诊疗的未来方向。
熊玉锋, 蔡贞, 李少川, 等. 生物信息学分析在病原微生物宏基因组高通量测序应用中的现状与挑战[J]. 中华医学杂志, 2023, 103(15): 1098-1102. DOI: 10.3760/cma.j.cn112137-20221208-02598.
所有作者均声明不存在利益冲突

























