
患儿为6岁7月男童,因“尿崩症原因待查”就诊于北京协和医院。患儿自幼多饮、多尿,近1年身高增速减慢,禁水试验和简易去氨加压素试验支持中枢性尿崩症诊断。垂体MRI未见明确占位性病变。基因检测发现AVP基因突变,符合遗传性中枢性尿崩症诊断。加用去氨加压素替代治疗后,患儿每日饮水量、尿量及夜尿次数均减少至正常水平。遗传性中枢性尿崩症罕见,自幼起病的中枢性尿崩症患者,在积极筛查常见病因的同时,应警惕遗传性尿崩症的可能性。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿男,6岁7月,主因“多尿、口干、多饮5年余”于2022年2月16日收入北京协和医院内分泌科。患儿为第一胎第一产,母亲妊娠3个月起出现多尿、口干、多饮等症状,饮水量3~4 L/d,未就诊,产后上述症状自行缓解;妊娠40周时因胎膜早破行剖宫产,患儿无产伤及窒息,出生身长52 cm,体重3 600 g。2017年初(患儿1岁半),家属发现患儿多尿、多饮,饮水量6~9 L/d,尿量与饮水量相当,尿色清亮如水,夜尿5~6次/晚,生长发育未见明显异常。自2021年初(患儿5岁半)始,患儿身高的增长速率较同龄儿减慢,近1年身高增加约3 cm,否认头痛、视力下降、视野缺损、恶心、呕吐和怕冷等症状。2022年1月20日于外院查尿比重为1.002;血生化结果:血钠:143 mmol/L,血钾:3.8 mmol/L,血糖:4.78 mmol/L,血钙:2.51 mmol/L;血常规结果:白细胞:11.79×109/L,中性粒细胞百分比:72.8%;垂体增强MRI:未见异常强化信号影,垂体后叶T1WI高信号显示模糊,垂体柄居中。为进一步诊治以“尿崩症原因待查”收入北京协和医院内分泌科。患儿自起病以来,精神、睡眠、食欲可,自幼大便次数较少,每3~5日排便一次,大便干结,小便如前所述,近5个月体重下降1.5 kg。
既往史:2岁时头顶部外伤,对症治疗后恢复良好。家族史:否认家族中有多尿、口干、多饮的病史,否认家族性垂体肿瘤、尿崩症病史。父亲身高176 cm,母亲身高147 cm。
体格检查:身高123 cm(位于同年龄同性别儿童第50~75百分位),体重24 kg(位于同年龄同性别儿童第50百分位),体质指数(BMI)为15.86 kg/m2,腰围55 cm,周身皮肤干燥,口唇略干,舌体湿润,腭弓高,未见内眦赘皮、低耳位、肘外翻、通贯掌和第四掌骨短等,心、肺、腹查体未见异常。阴毛Ⅰ期(Tanner分期),双侧睾丸体积约2 ml。
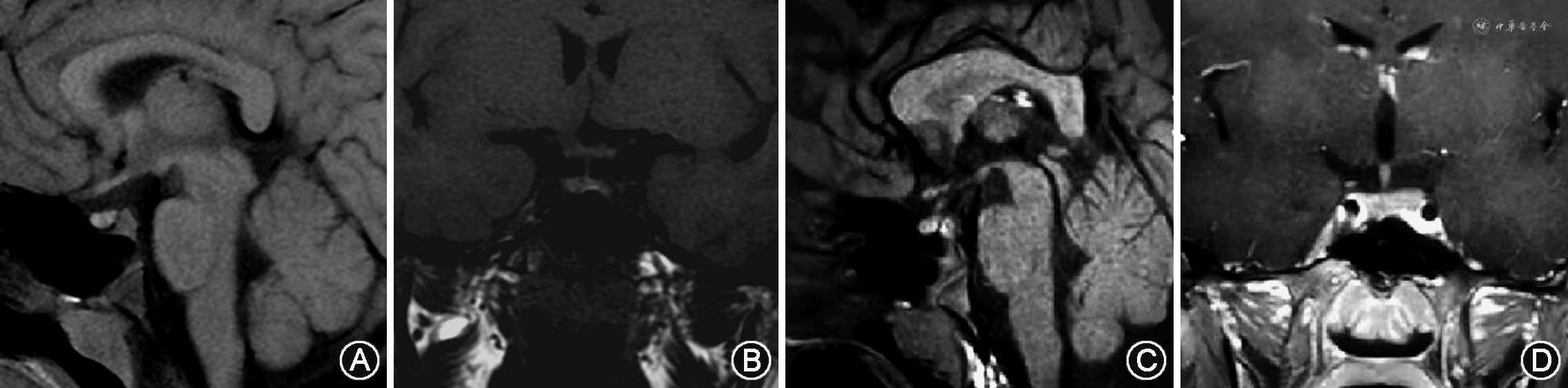
辅助检查:血生化结果:血钠:147 mmol/L,血氯:112 mmol/L,血糖、血钙正常;血常规结果:白细胞:6.10×109/L,中性粒细胞百分比:56.4%;垂体前叶功能评估提示生长激素轴、肾上腺激素轴、甲状腺激素轴功能大致正常,性腺轴符合青春期前的儿童水平(表1);禁水试验:禁水6 h后可见血渗透压升高至311 mOsm/kg、血钠升高至147 mmol/L,同时仍排出低渗尿,尿渗透压116 mOsm/kg、尿比重1.004,尿崩症定性诊断明确;简易去氨加压素试验:服用0.1 mg去氨加压素3 h后,尿量显著减少,尿渗透压升高至509 mOsm/kg,尿比重1.015,中枢性尿崩症诊断明确;尿崩症病因检查:抗核抗体谱:抗核抗体(ANA)低滴度(1∶80)阳性,核型为斑点型(S),其余自身抗体均为阴性;免疫球蛋白、补体、超敏C反应蛋白、血IgG亚类测定、甲胎蛋白、癌胚抗原、β人绒毛膜促性腺激素和血管紧张素转化酶等均正常;垂体增强MRI:鞍区、松果体、基底节区未见异常占位性病变或异常强化灶,垂体后叶短T1信号未见明显显示(图1);视野检查:无视野缺损;纯音测听及声导抗结果未见明显异常;骨龄相:骨龄5~6岁。
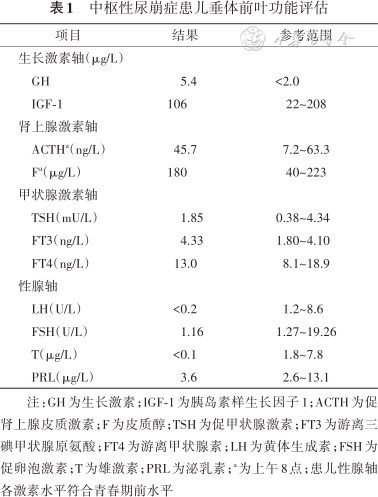
中枢性尿崩症患儿垂体前叶功能评估
中枢性尿崩症患儿垂体前叶功能评估
| 项目 | 结果 | 参考范围 |
|---|---|---|
| 生长激素轴(μg/L) | ||
| GH | 5.4 | <2.0 |
| IGF-1 | 106 | 22~208 |
| 肾上腺激素轴 | ||
| ACTHa(ng/L) | 45.7 | 7.2~63.3 |
| Fa(μg/L) | 180 | 40~223 |
| 甲状腺激素轴 | ||
| TSH(mU/L) | 1.85 | 0.38~4.34 |
| FT3(ng/L) | 4.33 | 1.80~4.10 |
| FT4(ng/L) | 13.0 | 8.1~18.9 |
| 性腺轴 | ||
| LH(U/L) | <0.2 | 1.2~8.6 |
| FSH(U/L) | 1.16 | 1.27~19.26 |
| T(μg/L) | <0.1 | 1.8~7.8 |
| PRL(μg/L) | 3.6 | 2.6~13.1 |
注:GH为生长激素;IGF-1为胰岛素样生长因子1;ACTH为促肾上腺皮质激素;F为皮质醇;TSH为促甲状腺激素;FT3为游离三碘甲状腺原氨酸;FT4为游离甲状腺素;LH为黄体生成素;FSH为促卵泡激素;T为雄激素;PRL为泌乳素;a为上午8点;患儿性腺轴各激素水平符合青春期前水平
诊疗经过:根据患儿病史,结合禁水试验及简易去氨加压素试验结果,考虑中枢性尿崩症诊断明确,加用去氨加压素代替治疗,并监测出入量、电解质、体重、血压和心率,嘱患儿调整饮水方式,量出为入。根据出入量调整去氨加压素用量至0.033 mg/次、3次/d,患儿全天入量可减少至3.5 L、尿量减少至1.8~2 L,夜尿减少至0次。患儿近一年生长缓慢,目前胰岛素样生长因子1(IGF-1)水平处于同龄儿均值,骨龄与实际年龄大致相仿,建议加用去氨加压素后监测生长速度,必要时进一步完善生长激素兴奋试验。
致病基因检测:患儿自幼起病,鞍区未见明确占位性病变,且常见中枢性尿崩症病因筛查未见明显异常,考虑遗传性中枢性尿崩症不除外。为明确诊断,对先证者及其父母行基因测序。先证者在编码精氨酸加压素(AVP)的AVP基因上发现1处杂合突变:c.232G>A(即:cDNA序列中第232位碱基G突变为A),突变类型为错义突变,导致氨基酸改变:p.Glu78Lys[即:蛋白质序列中第78位氨基酸由谷氨酸(Glu)突变为赖氨酸(Lys)]。先证者父母AVP基因均为野生型,不携带该突变基因,考虑先证者发生新生突变明确,而进一步全外显子测序未发现其他基因突变。查阅既往文献,可见该突变类型已有相关报告[1],因此考虑患者AVP突变导致的遗传性中枢性尿崩症诊断明确。
遗传性中枢性尿崩症为罕见疾病,且该患者家系成员中无尿崩症病史,临床医生诊断中枢性尿崩症病因时,容易遗漏遗传性因素。
由于夜间多尿、频繁起夜导致生长激素分泌的高峰被打断,尿崩症患儿可伴有生长发育迟缓的表现,临床医生应追问既往身高增长情况,并完善生长激素、IGF-1及骨龄检测,详细评估生长发育情况。
对于自幼起病的尿崩症患儿,积极筛查常见病因的同时,需警惕遗传性尿崩症的可能,并完善相关基因检测。
生长发育迟缓的中枢性尿崩症患儿经激素替代治疗后,身高体重可迅速追赶同龄儿童,因此应尽早积极治疗,以免错失关键生长发育时机造成永久性生长落后。
本例患儿幼年男童,慢性病程,自幼出现多饮、口干、多尿,饮水量多达9 L/d,尿量与饮水量相当,多尿诊断明确。多尿的病因主要包括渗透性利尿、精神性多饮及尿崩症。该患儿血糖水平正常,无甘露醇等高渗药物使用史,排除渗透性利尿可能;日、夜尿量均显著增多,夜尿5~6次/晚,且无焦虑、抑郁等精神因素,考虑精神性多饮可能性小;持续排出大量低渗尿,尿比重明显减低,考虑尿崩症可能性大。患儿禁水试验后血渗透压、血钠明显升高,同时尿渗透压、尿比重仍明显减低,尿崩症定性诊断明确;服用去氨加压素后,尿量显著减少,尿渗透压升高,表明对外源性AVP有反应,中枢性尿崩症定位诊断明确。此外,患儿垂体增强MRI提示垂体后叶短T1信号未见明确显示,表明垂体后叶缺乏储存的AVP,进一步支持中枢性尿崩症诊断。
在中枢性尿崩症病因筛查方面,肿瘤性疾病是首要常见病因。患儿无头痛、视力下降、视野缺损等颅内占位表现,垂体MRI显示未见鞍区异常占位性病变或异常强化灶,且血清肿瘤标志物未见异常,肿瘤性疾病证据不足;病程中无发热等感染表现。幼年男童非自身免疫性疾病好发人群,血清炎症指标、免疫球蛋白、补体、IgG亚类测定均正常,无感染性炎症、自身免疫性垂体炎相关证据;完善多部位超声未发现其他器官系统受累情况,考虑组织细胞病可能性小;垂体MRI显示垂体柄完整,排除垂体柄阻断综合征可能。基因检测发现患儿AVP基因出现一处错义突变,为杂合突变,其父母均不携带该突变基因,故遗传性中枢性尿崩症诊断明确。患儿母亲AVP基因为野生型,但在孕期表现出多尿症状,考虑是由于胎盘合成AVP酶,加速孕妇血液循环中的AVP降解所导致[2],产后相关症状消失也支持上述解释。
尿崩症是一种发病率约为1/25 000的罕见疾病,其中病因为遗传性因素者更为罕见[3, 4, 5]。国外一项对138例中枢性尿崩症患儿的回顾性研究发现,仅有2例患儿的病因为遗传性因素[6]。大多数遗传性中枢性尿崩症与AVP基因突变相关,为常染色体显性遗传[7],目前人类基因突变数据库中已报道的AVP基因致病突变有87个[8];此外,还有少数病例携带有WFS1基因突变,为常染色体隐性遗传[9]。目前文献报道的AVP基因突变中,大部分为错义突变和无义突变,少见缺失突变和剪接位点突变[10]。AVP基因编码三种最终产物,包括AVP、后叶激素运载蛋白Ⅱ(NPⅡ)及和肽素,NPⅡ在AVP的加工、运输和分泌中发挥重要作用[11]。
本例患儿出现的错义突变位于NPⅡ编码区,干扰了NPⅡ与AVP间蛋白质的相互作用,进而影响AVP向垂体后叶运输和分泌。AVP前蛋白无法完成正确折叠和加工,因而积累在内质网中,对神经元产生毒性损伤引发凋亡,进一步加重了AVP缺乏[12]。
尿崩症常影响儿童睡眠、食欲及生长发育,若长期得不到治疗,部分患儿会出现身高偏矮、生长发育迟缓的表现。一项在3个遗传性中枢性尿崩症家系中开展的临床研究发现,携带AVP突变的患儿均表现出不同程度的生长发育迟滞,进行激素替代治疗后其身高体重均达到同龄儿的均值[13]。本例患儿IGF-1水平处于同龄儿均值,且骨龄与实际年龄相当,考虑近期生长缓慢与患儿夜间多尿、频繁起夜及打断夜间生长激素分泌的高峰关系较大。拟给予尿崩症对症治疗后,观察其生长速度改善情况,再决定是否行生长激素兴奋试验进一步评估生长激素轴功能。
龚心悦, 李冉, 朱伊祎, 等. 第573例 男童—多尿—多饮—身高发育迟缓[J]. 中华医学杂志, 2023, 103(15): 1154-1156. DOI: 10.3760/cma.j.cn112137-20220928-02049.
所有作者均声明不存在利益冲突

























