
探讨原发性周围神经过度兴奋综合征(PNHS)患者的临床和神经电生理特征。
回顾性纳入2016年4月至2023年1月就诊于首都医科大学附属北京天坛医院的原发性PNHS患者20例。记录患者的临床表现及神经电生理检测结果。依据血和脑脊液抗结旁区接触蛋白相关蛋白-2(CASPR2)和(或)抗富亮氨酸胶质瘤失活1蛋白(LGI1)抗体检测结果,比较抗体阳性组和抗体阴性组患者的临床和电生理特征。
患者男12例,女8例,年龄(44.0±17.2)岁,病程[M(Q1,Q3)]2.3(1.1,11.5)个月。运动症状包括肌束颤动、肌颤搐和肌肉酸痛,肌肉痉挛和僵硬;以下肢(17例)多见,其次是上肢(11例)、头面部(11例)和躯干(9例)。19例(19/20)患者存在感觉异常和(或)自主神经障碍。13例患者出现中枢神经系统症状。5例患者合并肺癌或胸腺病变。针极肌电图特征性异常自发电位包括肌颤搐电位(19例)、束颤电位(12例)、痉挛电位(3例)、神经性肌强直电位(1例)等,以下肢肌肉、特别是腓肠肌(12例)多见。8例患者存在后发放电位,7例出现胫神经后发放。7例患者血清抗CASPR2抗体阳性,其中3例同时存在抗LGI1抗体;1例患者单纯血清抗LGI1抗体阳性。与抗体阴性组相比(n=12),抗体阳性组(n=8)患者的病程更短[M(Q1,Q3),1.8(1,2)个月比9.5(3.3,20.3)个月,P=0.012],胫神经后发放电位的出现率更高(6/8 比 2/12,P=0.019)。抗体阳性患者免疫治疗方案(多药、单药、未免疫治疗分别为6、2、0例)与抗体阴性组(3、6、3例)比较差异有统计学意义(U=21.00,P=0.023)。
原发性PNHS患者运动神经兴奋症状、针极肌电图自发放电和M波后发放电位均以下肢多见;临床应重视对感觉神经和自主神经过度兴奋的表现的识别。抗CASPR2抗体阳性的PNHS患者可能需要多种药物联合免疫治疗。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
周围神经过度兴奋综合征(peripheral nerve hyperexcitability syndrome,PNHS)是一组罕见的神经系统疾病,其临床特征为自发性肌肉活动,表现为肌束颤动、肌颤搐、痉挛、僵硬等,针极肌电图存在束颤电位、肌颤搐电位、神经性肌强直电位等运动单位自发放电[1]。另一个神经电生理特征是后发放,即低频重复刺激运动神经产生初始复合肌肉动作电位后的持续性发放,可作为运动神经过度兴奋的敏感标志[2, 3]。后天获得性PNHS可分为原发性和继发性两类,原发性PNHS包括痉挛-束颤综合征、Isaacs综合征和Morvan综合征;后者可继发于运动神经元病、放射性和中毒性周围神经病等[4]。
自身免疫机制在原发性PNHS的病理生理中发挥重要作用,主要通过干扰电压门控钾离子通道(voltage-gated potassium channel,VGKC)的功能而致病[1]。抗结旁区接触蛋白相关蛋白-2(contactin-associated protein2,CASPR2)和富亮氨酸胶质瘤失活1蛋白(leucine-rich glioma-inactivated1,LGI1)是抗VGKC复合物抗体的主要靶抗原[5, 6]。一部分PNHS与肿瘤/副肿瘤性疾病,特别胸腺瘤相关[4]。该综合征目前国内报道的病例较少,临床症状有时不典型,易被临床医生忽视或误诊。本研究旨在归纳总结原发性PNHS的临床和电生理特征,为临床诊治提供依据。
回顾性收集首都医科大学附属北京天坛医院2016年4月至2023年1月收治的PNHS患者,检索出院诊断中包含“周围神经过度兴奋综合征”或“神经性肌强直”或“Morvan综合征”或“痉挛-束颤综合征”的患者共23例。排除1例早期诊断为神经性肌强直,最终证实为运动神经元病的患者,2例遗传性PNHS患者,最终纳入原发性PNHS患者20例。本研究符合《赫尔辛基宣言》对伦理的要求,患者知情同意豁免。
1.资料收集:收集患者的临床信息,包括性别、年龄、病程,记录完整的病史,周围神经系统及中枢神经系统症状及分布,神经系统体格检查。
2.神经电生理检查:所有患者行运动和感觉神经传导测定、F波、针极肌电图检查,记录M/F波后发放的出现率,针极肌电图自发电位的类型及出现的部位。依据患者的临床症状和体征,部分患者进行皮肤交感反应(sympathetic skin response,SSR)、心率变异率检测。检测方法和正常值参照首都医科大学附属北京天坛医院肌电图室自行建立标准。
3.实验室检查:入院后行血常规、生化、免疫学指标等常规检测。采用基于细胞底物的实验方法对所有患者进行血清/脑脊液抗CASPR2和抗LGI1抗体检测(间接免疫荧光法,抗体滴度≥1∶10定义为阳性),部分患者同时完善了血清抗VGKC复合物抗体检测(放免法,正常范围0~72 pmol/L)。依据患者的症状和体征,选择性完善头颅磁共振、脑电图、多导睡眠图检查。所有患者进行肿瘤筛查,包括血清肿瘤标记物、副肿瘤综合征相关抗体、胸腹部和盆腔CT,女性患者行乳腺和妇科超声检查;当高度怀疑恶性肿瘤时,进行正电子发射断层扫描(PET-CT)。
4.治疗和随访:记录患者的治疗方案,包括免疫治疗、对症治疗、肿瘤等合并症的治疗等。所有患者出院后通过再住院、门诊或电话随访至少一次。
采用SPSS25.0软件建立数据库,并对数据进行描述性统计分析。符合正态分布和方差齐性的计量资料用表示,组间比较采用两独立样本t检验;不符合正态分布的计量资料用M(Q1,Q3)表示,组间比较采用曼-惠特尼U检验;计数资料用例数或例次表示,组间比较采用Fisher精确检验。双侧检验,检验水准α=0.05。
患者男12例,女8例。患者的临床特征及治疗转归情况见附表1(扫描本文首页二维码查阅)。患者年龄10~71(44±17)岁,病程2.3(1.1,11.5)个月。运动神经兴奋的症状包括肌束颤动(17/20)、肌颤搐(17/20)和肌肉酸痛(12/20),肌肉痉挛(3/20)和僵硬(4/20)。上述症状以下肢多见(17/20),其次是上肢(11/20)、头面部(11/20)和躯干(9/20)。3例患者症状只局限在下肢,另3例患者只局限在头面部。

17例患者出现自主神经功能障碍,其中最多见的症状是多汗(16/20)和尿便障碍(10/20),少数患者可出现体位性低血压(5/20)和心动过速(3/20)。16例患者有感觉异常的主诉,包括肢体皮肤麻木、蚁走感、瘙痒、烧灼感、过电感、发冷感。13例患者出现中枢神经系统症状,包括失眠(9/20)、记忆力减退(8/20)、焦虑/抑郁(7/20)、幻觉(1/20)、性格改变(1/20)和癫痫(1/20);所有患者进行头颅磁共振检查,2例患者显示双侧海马、颞叶T2/FLAIR高信号,其余患者均未见明显异常。
9例患者因同时存在周围神经过度兴奋、自主神经功能障碍和睡眠障碍等中枢神经系统症状,被诊断为Morvan综合征;其中1例患者上呼吸道感染2周后出现四肢远端为著的肢体麻木无力、肌颤搐、幻觉、睡眠障碍等,考虑Morvan综合征合并多发性感觉运动神经病,血清和脑脊液抗CASPR2抗体均阳性,最终诊断为抗CASPR2抗体相关神经系统病变。另11例患者被诊断为Issacs综合征。
所有患者进行了肿瘤筛查,包括血清肿瘤标志物和胸腹盆CT扫描,其中3例接受了PET-CT。检查发现1例患者患肺癌,因病变体积小(直径6 mm),肿瘤科建议密切随访,具体类型未定性;3例患者合并胸腺增生;1例患者发病前半年发现胸腺瘤,行胸腺切除术及术后化疗4疗程;2例血清抗Yo抗体阳性,但PET-CT未发现明确恶性病变。
19例(19/20)患者针极肌电图具有异常自发放电(附表2,扫描本文首页二维码查阅)。19例患者出现肌颤搐电位,呈双联、三联或多联发放;12例患者出现束颤电位;3例患者出现痉挛电位;1例患者出现神经性肌强直电位;异常自发电位以下肢多见(12/20),其中腓肠肌(12/15);其次是上肢肌(8/20)和面肌(8/20)。仅在1例合并多发性周围神经病的患者上下肢所检肌肉和1例合并腰椎病的患者、单侧胫前肌存在正锐波和纤颤电位;前者上下肢所检肌肉运动单位动作电位(motor unit action potential,MUAP)及募集电位呈神经源性损害,其余患者均未见明显异常。
只有2例患者出现了神经传导检测异常:1例合并多发性周围神经损害,上、下肢感觉、运动神经传导速度均下降(正中神经运动神经传导速度 30 m/s),复合肌肉动作电位(compound muscle action potential,CMAP)及感觉神经动作电位(sensory nerve action potential,SNAP)波幅均降低(正中神经CMAP波幅0.35 mV);另1例合并腕管综合征,双侧正中神经感觉、运动传导速度减慢,右侧正中神经 SNAP波幅下降。8例患者存在后发放电位,正中神经、尺神经、胫神经的后发放电位阳性率分别为3/7、4/16和7/19。此外,1例以面部肌肉抽动为主要表现的患者,双侧面神经F波检测也可以看到后发放。
自主神经功能检测结果显示,7例(7/15)患者上、下肢SSR异常,2例波形未引出,5例SSR波幅下降[上肢波幅0.36(0.18,0.64)mV,下肢波幅0.19(0.06,0.36)mV],提示交感神经功能障碍;5例(5/13)患者心率变异率异常,提示副交感神经功能障碍。
所有患者进行血清和脑脊液抗CASPR2、LGI1抗体检测。7例患者血清抗CASPR2抗体阳性;其中3例患者同时存在血清抗LGI1抗体阳性,其抗CASPR2抗体的滴度均高于抗LGI1抗体的滴度;仅1例患者血清和脑脊液抗CASPR2抗体均阳性,该患者临床表现为多发性感觉运动神经病、周围神经过度兴奋和边缘叶脑炎。1例合并肺癌的患者,呈血清抗LGI1抗体单阳性。
4例患者同时进行了血清抗VGKC复合物抗体检测,其中2例患者血清抗CASPR2和(或)LGI1抗体阳性,其抗VGKC复合物抗体检测结果呈阳性;2例患者血清抗CASPR2/LGI1抗体均为阴性,抗VGKC复合物抗体检测亦呈阴性。
所有患者进行了血清自身抗体谱筛查,只有1例合并胸腺瘤的患者,发现抗核抗体(antinuclear antibodies,ANA)1∶100核颗粒型,抗横纹肌抗体阳性,抗Ro-52抗体弱阳性,但无系统性结缔组织病的证据。
17例患者接受免疫治疗,8例应用IVIG联合激素治疗,其中1例加用利妥昔单抗治疗;7例IVIG治疗;2例激素治疗。2例患者因经济因素及担心药物不良反应拒绝免疫治疗,1例患者应用对症治疗后症状好转,未再进一步加用免疫治疗。
其他治疗药物包括奥卡西平(6/20)、卡马西平(5/20)、加巴喷丁(7/20)、氯硝西泮(11/20)、度洛西汀(5/20)、文拉法辛(2/20)、普瑞巴林(2/20)、苯妥英钠(1/20)等。1例合并肺癌及3例胸腺增生患者均未行手术治疗。对所有患者进行随访,18例(18/20)患者均有不同程度好转,1例患者症状稳定无明显改善或加重,1例失访。
依据抗CASPR2、LGI1抗体检测结果,将患者分为抗体阳性组(n=8)和抗体阴性组(n=12)。两组患者的临床和神经电生理特征如表1所示。两组患者的性别、年龄差异均无统计学意义。抗体阳性组患者的病程显著短于抗体阴性组;运动症状累及的范围更广、中枢神经系统症状更多见,但差异无统计学意义。抗体阳性组患者神经传导M波后发放的出现率显著高于抗体阴性组,特别是在胫神经。抗体阳性组所有患者接受免疫治疗,且6/8患者需要丙球、激素(或利妥昔单抗)多药联合治疗,2/8单药治疗;抗体阴性组中3/12患者经多药联合治疗,6/12单药治疗,3/12仅对症治疗(U=21.00,P=0.023)。
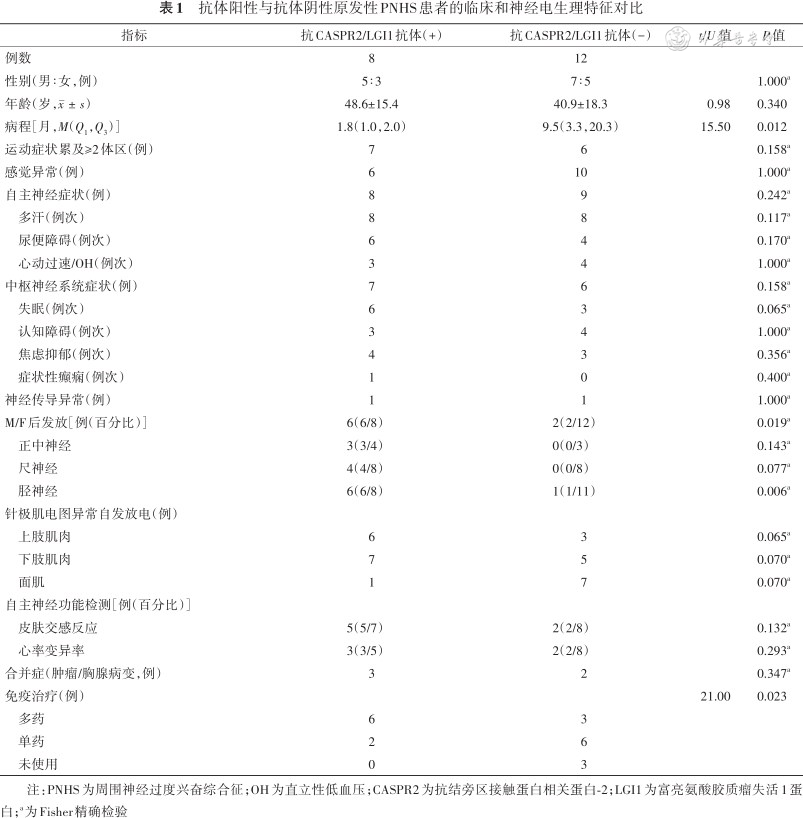
抗体阳性与抗体阴性原发性PNHS患者的临床和神经电生理特征对比
抗体阳性与抗体阴性原发性PNHS患者的临床和神经电生理特征对比
| 指标 | 抗CASPR2/LGI1抗体(+) | 抗CASPR2/LGI1抗体(-) | t/U 值 | P值 |
|---|---|---|---|---|
| 例数 | 8 | 12 | ||
| 性别(男∶女,例) | 5∶3 | 7∶5 | 1.000a | |
| 年龄(岁,) | 48.6±15.4 | 40.9±18.3 | 0.98 | 0.340 |
| 病程[月,M(Q1,Q3)] | 1.8(1.0,2.0) | 9.5(3.3,20.3) | 15.50 | 0.012 |
| 运动症状累及≥2体区(例) | 7 | 6 | 0.158a | |
| 感觉异常(例) | 6 | 10 | 1.000a | |
| 自主神经症状(例) | 8 | 9 | 0.242a | |
| 多汗(例次) | 8 | 8 | 0.117a | |
| 尿便障碍(例次) | 6 | 4 | 0.170a | |
| 心动过速/OH(例次) | 3 | 4 | 1.000a | |
| 中枢神经系统症状(例) | 7 | 6 | 0.158a | |
| 失眠(例次) | 6 | 3 | 0.065a | |
| 认知障碍(例次) | 3 | 4 | 1.000a | |
| 焦虑抑郁(例次) | 4 | 3 | 0.356a | |
| 症状性癫痫(例次) | 1 | 0 | 0.400a | |
| 神经传导异常(例) | 1 | 1 | 1.000a | |
| M/F后发放[例(百分比)] | 6(6/8) | 2(2/12) | 0.019a | |
| 正中神经 | 3(3/4) | 0(0/3) | 0.143a | |
| 尺神经 | 4(4/8) | 0(0/8) | 0.077a | |
| 胫神经 | 6(6/8) | 1(1/11) | 0.006a | |
| 针极肌电图异常自发放电(例) | ||||
| 上肢肌肉 | 6 | 3 | 0.065a | |
| 下肢肌肉 | 7 | 5 | 0.070a | |
| 面肌 | 1 | 7 | 0.070a | |
| 自主神经功能检测[例(百分比)] | ||||
| 皮肤交感反应 | 5(5/7) | 2(2/8) | 0.132a | |
| 心率变异率 | 3(3/5) | 2(2/8) | 0.293a | |
| 合并症(肿瘤/胸腺病变,例) | 3 | 2 | 0.347a | |
| 免疫治疗(例) | 21.00 | 0.023 | ||
| 多药 | 6 | 3 | ||
| 单药 | 2 | 6 | ||
| 未使用 | 0 | 3 |
注:PNHS为周围神经过度兴奋综合征;OH为直立性低血压;CASPR2为抗结旁区接触蛋白相关蛋白-2;LGI1为富亮氨酸胶质瘤失活1蛋白;a为Fisher精确检验
PNHS患者特征性表现是肌肉自发性活动,本研究中所有患者均有不同形式的运动神经兴奋症状,以肌束颤动和肌颤搐最常见,多见于下肢。除四肢和躯干外,3/20的患者的症状仅局限在头面部,面神经F波检测也可见后发放电位,有助于临床诊断。既往有文献报道临床表现为孤立性感觉异常或多汗,经神经电生理检查确诊的PNHS[7, 8]。本研究中80%患者存在感觉异常,表现为肢体皮肤麻木、蚁走感、瘙痒、烧灼感、过电感等刺激性感觉症状;17/20的患者存在自主神经兴奋,表现为多汗、尿便障碍、心血管功能紊乱等。Hart等[1]报道35%自身免疫性PNH患者有感觉症状;李杨等[9]汇总既往国内外报道的Morvan综合征出现自主神经功能障碍表现的患者占65%以上。因此,感觉和自主神经受累在PNHS并不少见。
针极肌电图异常自发放电中,肌颤搐电位(19/20)出现率最高。Maddison等[10]和Wu等[11]的研究中肌颤搐电位出现率(分别为11/11、14/19)也明显高于其他类型自发电位。有研究显示,在肢体远端肌肉记录到的自发电位多于肢体近段肌肉[1],自发放电的频率可能与支配肌肉的神经长度呈正相关[10]。本研究中自发电位在下肢远端肌肉——腓肠肌最为常见;胫神经后发放电位的检出率也高于正中神经和尺神经。
CASPR2 在中枢神经和周围神经有髓纤维轴突的郎飞结近结旁区表达,抗CASPR2抗体阳性患者可出现认知障碍、癫痫发作、边缘叶脑炎、小脑性共济失调、神经痛等症状[12],其中PNHS的出现率为37.7%[13];而PNHS患者中抗CASPR2抗体阳性的比例为31.6%[14],与本研究中的检出率(7/20)相近。由于CASPR2抗体主要在外周产生,测定血清抗体比脑脊液更敏感[15]。本研究中6例患者单纯血清抗体阳性;1例患者血清和脑脊液抗体均阳性,血清滴度高于脑脊液滴度,该患者除PNHS、边缘叶脑炎外,同时存在急性多发性感觉运动神经病。周围神经脱髓鞘和(或)轴索损害在CASPR2抗体阳性患者较为罕见,Tan等[16]汇总了既往文献报道的8例类似患者,其中6例表现为吉兰-巴雷综合征(Guillain-Barre syndrome,GBS),2例表现为慢性炎症性脱髓鞘性多发性神经病(chronic inflammatory demyelinating polyneuropathy,CIDP),并提出CASPR2抗体可能通过改变VGKC的聚集和功能,导致有髓神经纤维的轴突损伤,但不能排除其他共存的尚未确定的神经元抗体的作用。
LGI1主要在海马区和颞叶皮质表达,抗LGI1抗体阳性患者常表现为面臂肌张力障碍和边缘叶脑炎,较少表现为PNHS[17]。本研究中只有1例PNHS患者单纯抗LGI1抗体阳性,LGI抗体与PNHS的关系尚有待进一步确认。相比之下,LGI1抗体伴随CASPR2抗体阳性较多见[17]。
比较抗体阴性和抗体阳性患者的临床和电生理特征,抗体阳性患者的病程更短,后发放电位出现率更高。无论是否存在抗VGKC复合物抗体,PNHS患者都可从免疫治疗中获益。两组患者免疫治疗的强度(多药联合、单药治疗、未经免疫治疗的比例)差异存在统计学意义,提示抗体阳性患者可能更多需要联合免疫治疗。但抗体阴性组有2例因个人因素拒绝使用免疫治疗,这一结果有待在更多患者中进一步验证。
综上所述,PNHS患者运动神经兴奋的症状、特征性肌电图自发放电和后发放电位均以下肢最多见;临床需重视对感觉和自主神经过度兴奋的症状的识别。血清抗CASPR2抗体阳性的PNHS患者可能需要多种药物联合免疫治疗。本研究存在一定的局限性。首先,本研究为回顾性研究,神经电生理检查所检神经和肌肉的选择受临床症状的影响,例如1例症状仅局限在面部的患者只做了面肌肌电图。其次,抗体阳性的例数较少,部分患者未提供CASPR2抗体的滴度,未进一步分析抗体滴度与疾病严重程度之间的关系。
邰宏飞, 陈彬, 牛松涛, 等. 原发性周围神经过度兴奋综合征的临床及神经电生理特征[J]. 中华医学杂志, 2023, 103(25): 1925-1930. DOI: 10.3760/cma.j.cn112137-20230303-00320.
所有作者声明不存在利益冲突

























