
本研究收集2个汉族Okihiro综合征家系患者的临床表现,应用外显子组测序对2个家系的先证者进行检测,Sanger测序对家系成员进行基因型分析,以确定生物学发病机制,并为家系中的3个高危胎儿提供产前诊断。2个家系的患者表现出广泛的表型,尤其是家系1的先证者在胎儿期就发现超声异常(桡骨畸形、姿势异常、永存动脉干和室间隔缺损等心脏畸形),其余患者表现从严重表型(前臂严重缩短和变形、杜安异常、听力障碍),到不太明显的情况(仅有大鱼际发育不良、桡骨茎突较短小)。外显子组测序在2个家系的先证者中分别发现SALL4基因变异:c.844delC p.(Q282Kfs*8)和c.2210delG p.(G737Vfs*23),其中c.2210delG p.(G737Vfs*23)为新变异。家系验证发现患者成员均携带变异,正常成员中均未检测到。同时,3个高危胎儿中共检出1例正常基因型胎儿,2例携带杂合变异胎儿并终止妊娠。本研究通过外显子组测序和Sanger测序明确了2个Okihiro综合征家系的分子病理学原因,丰富了Okihiro综合征在围产期的临床表现和SALL4基因突变图谱,同时为3个高危胎儿提供产前诊断,有效降低生育患儿的风险。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Okihiro综合征也称为杜安射线综合征或肢端-肾-眼综合征,是一种罕见的临床表现广泛的常染色体显性遗传疾病,典型临床表现是上肢畸形(从三指拇指到单侧或双侧桡骨缺失),和眼病畸形(杜安异常)[1, 2]。杜安异常主要包括眼睛外展受限,眼睑裂狭窄,内收时眼球收缩[3]。尽管目前认为上肢桡骨缺损和杜安异常是该疾病的主要特征,但它们仅分别出现在91%和65%的病例中[4],在家族内部和家族之间是极其可变的。其他常见的表型包括心脏异常、肾脏异常、脊椎异常等,不太常见的特征还有感音神经性耳聋、肛门闭锁和色素障碍[1,5]。目前Okihiro综合征的发病率未知,而且国内鲜有Okihiro综合征相关报道。在2002年,Al-Baradie等[1]和Becker等[6]均发现SALL4基因变异引起Okihiro综合征。SALL4基因位于染色体20q13.13-q13.2,包含4个外显子,编码1 053个氨基酸组成具有8个C2H2型锌指基序(ZFM)的锌指蛋白转录因子。SALL4在胚胎发育早期广泛表达,同时也是许多肿瘤的关键基因,负责调节胚胎干细胞的自我更新,在胚胎发育过程中起重要作用。本研究通过对2个中国汉族Okihiro综合征家系进行临床资料分析及基因变异检测,旨在完善临床表现尤其围产期的临床表现和发现致病基因变异,同时避免受累患儿出生为2个家系提供产前诊断。
2个无亲缘关系的中国汉族Okihiro综合征家系均因“上肢畸形,姿势异常”就诊于郑州大学第一附属医院遗传产前诊断中心,回顾每个先证者的家族史、病史。经过签署知情同意书后,收集2个Okihiro综合征家系的家系成员临床资料和外周血样本。本研究经郑州大学第一附属医院医学伦理委员会批准(批号:KS-2018-KY-36)。
1.基因组DNA的提取:抽取家系成员的外周静脉血2 ml,乙二胺四乙酸抗凝。于妊娠11~14周的胎儿在超声引导下经腹抽取胎儿绒毛组织进行产前基因诊断。应用Omega DNA提取试剂盒(美国Omega公司)提取DNA。
2.外显子组测序(ES)及分析:应用ES对2个家系中的先证者进行检测,使用NexteraTMFlex for Enrichment试剂盒(美国Illumina公司)捕获进行文库构建,应用150 bp双端测序试剂盒在NovaSeq 6000(美国Illumina公司)测序平台测序,测序平均深度200×,大于20×的靶向reads达到97%以上。测序数据与hg19数据库比对获取变异文件,上传变异数据到EGIS亿解基因数据解读平台(上海翰垚生物医学公司)进行变异注释和筛选,筛选要求:(1)根据上肢骨骼形态异常(HP:0040070)获得422个OMIM致病基因;(2)筛选出外显子和剪切区检测到的变异;(3)与千人数据库、NCBI dbSNP数据库、ExAC数据库、ESP6500 基因组数据库和gnomAD_exome数据库等进行比对,排除等位频率在1%以上的已知多态性位点。筛选得到的变异依据美国医学遗传学与基因组学学会(ACMG)遗传变异分类标准与指南进行变异评级。
3.PCR及Sanger测序:应用Gene tool软件针对2个可疑突变位点进行引物设计(表1),并在ABl3130xl基因测序仪(美国ABI公司)上对产物直接进行双向测序。
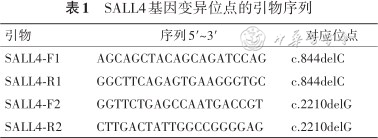
SALL4基因变异位点的引物序列
SALL4基因变异位点的引物序列
| 引物 | 序列5′~3′ | 对应位点 |
|---|---|---|
| SALL4-F1 | AGCAGCTACAGCAGATCCAG | c.844delC |
| SALL4-R1 | GGCTTCAGAGTGAAGGGTGC | c.844delC |
| SALL4-F2 | GGTTCTGAGCCAATGACCGT | c.2210delG |
| SALL4-R2 | CTTGACTATTGGCCGGGGAG | c.2210delG |
4.产前诊断和随访:2个家系中的3个高危胎儿在孕11~14周进行绒毛穿刺术获得产前诊断绒毛样本,应用美国Promega公司的PowerPlex 16 HS System试剂盒在GeneMapper v3.2软件进行母体污染排除实验分析。应用PCR结合Sanger测序的方法,对高危胎儿进行相应位点的检测。3个高危胎儿娩出后留取脐带血进行基因诊断,并进行电话随访。
1.家系1(图1A):先证者(Ⅲ:4)因在孕期超声发现左手形态异常,经人类基因组拷贝数变异检测未见异常而就诊。孕12+4周的B超显示左侧桡骨缺失伴手姿势异常,右手姿势呈现垂腕状;由于孕周小而心内结构显示模糊未见明确异常。胎儿母亲有类似情况的孕产史,受累胎儿Ⅲ:3在孕22周超声影像显示头颅、胸腔、脊柱正常,但右侧桡骨缺失伴右手姿势异常,且超声心动图显示胎儿心脏畸形,室间隔上部回声连续性中断,宽约3.4 mm,左心室可见一个大小约为2.5 mm×1.9 mm强光点,仅见单一大动脉起自左右心室间,跨与室间隔之上,位于气管的左侧,左右肺动脉似起源于大动脉根部,走行迂曲,左右肺动脉内径1.4~1.5 mm;下腹部肠管回声增强,范围约27 mm×16 mm。
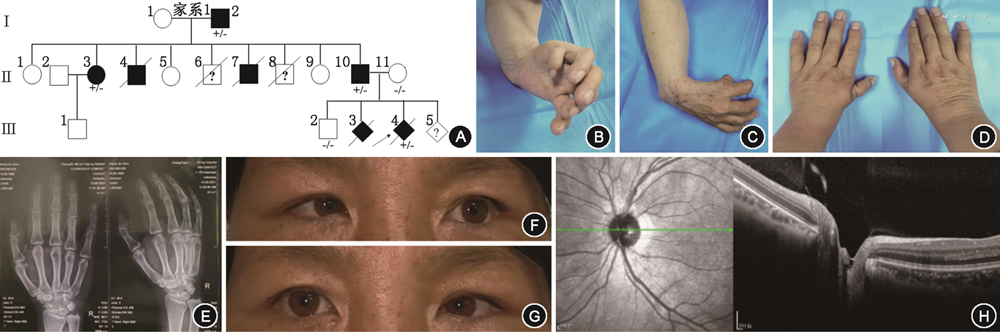
家系中其他3个患者临床表现差异很大:(1)骨骼系统:患者I:2双上肢桡侧球棒手畸形,前臂短小弯曲,桡骨缺失,手指与尺骨纵轴成角畸形,双手拇指缺如并且姿势异常(图1B、1C);患者Ⅱ:3大拇指发育异常,掌指关节不稳,大鱼际明显发育不良(图1D);患者Ⅱ:10双侧拇指未见明显异常,X射线显示双侧桡骨茎突较短小,尺骨关节面超过桡骨关节面,双侧舟骨及大多数角骨体积小,桡腕关节间隙变窄,提示双侧桡骨远端发育不良(图1E)。(2)眼睛系统:患者I:2眼睛未见异常;患者Ⅱ:3明显的杜安异常,双眼外展受限(图1F、1G);患者Ⅱ:10眼睛检查发现视盘发育不良(图1H),中央点状白内障。(3)其他系统,患者Ⅱ:3左耳听力受损,心脏超声检查3个患者(I:2,Ⅱ:3,Ⅱ:10)均未见明显异常。
2.家系2(图2A):先证者(Ⅱ:5),女,48岁,因双侧上肢畸形姿势异常而就诊,X射线显示双侧前臂短小屈曲桡骨缺如,双手拇指缺如,余指屈曲(图2B、2C)。家系中其他患者骨骼畸形症状相对较轻,患者(Ⅲ:7)为多指且手术矫正后,其余患者的双手存在鱼际发育不良(图2D、2E)。受累胎儿(Ⅳ:2)在孕18周超声影像结果:头颅、脊柱、四肢均显示正常,右手大拇指呈背曲状态;右肾区未检测到肾脏回声,在盆腔内膀胱后方可见疑似肾脏样回声,大小约14 mm×8 mm,提示胎儿疑似存在盆腔异位肾。该胎儿在孕22周超声复查,右手大拇指呈背曲状态,盆腔内膀胱后可见大小约22 mm×13 mm的疑似肾脏样回声,同时超声心动图显示胎儿心内结果正常。
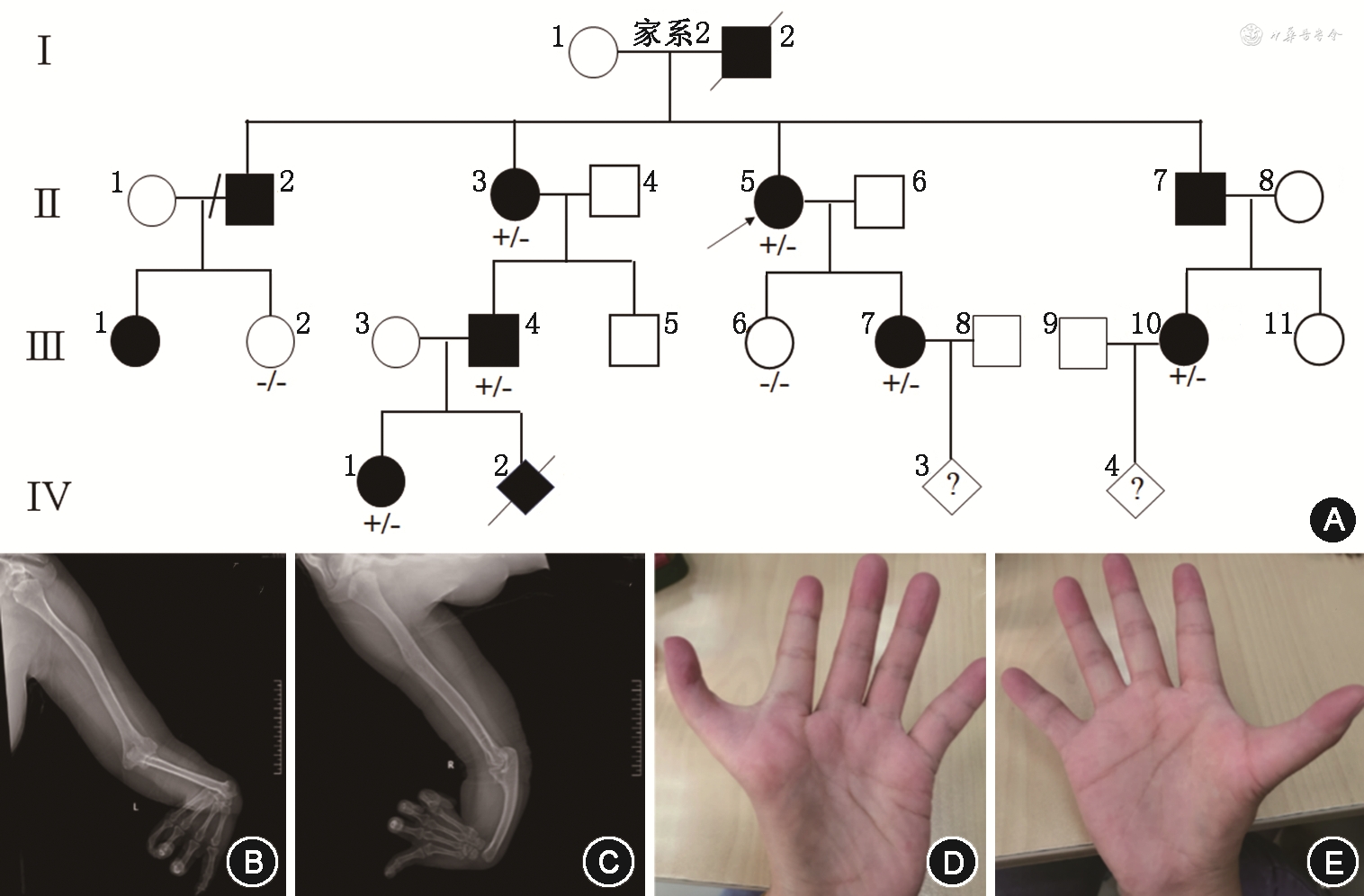
应用ES对2个先证者进行检测,经过数据筛选和分析,在家系1和家系2的先证者分别发现SALL4基因(NM_020436)c.844delC和c.2210delG杂合变异,预测分析分别产生290个氨基酸p.Q282Kfs*8和760个氨基酸p.G737Vfs*23的截段蛋白(表2)。2个变异在千人数据库、NCBI dbSNP数据库、ExAC数据库、ESP6500 基因组数据库和gnomAD_exome数据库等均未发现,其中c.2210delG为未经报道的新变异。应用Sanger测序进行家系验证,家系1患者成员(Ι:2,Ⅱ:3,Ⅱ:10,Ⅲ:4)均携带c.844delC p.(Q282Kfs*8)杂合变异,正常表型成员(Ⅱ:11,Ⅲ:2)则不携带;家系2患者成员(Ⅱ:3,Ⅱ:5,Ⅲ:4,Ⅲ:7,Ⅲ:10,Ⅳ:1)均携带c.2210delG p.(G737Vfs*23)杂合变异,正常表型成员(Ⅲ:2,Ⅲ:6)未发现该变异。根据ACMG指南,2个变异均判定为致病性变异(PVS1+PM2_S+PP1+PP4)。

外显子组测序检测Okihiro综合征家系先证者的SALL4基因致病变异位点
外显子组测序检测Okihiro综合征家系先证者的SALL4基因致病变异位点
| 先证者 | 基因 | 转录本 | 外显子 | 核苷酸变化 | 氨基酸变化 | 类型 | 频率 | 变异说明 |
|---|---|---|---|---|---|---|---|---|
| 家系1 | SALL4 | NM_020436 | exon2 | c.844delC | p.Q282Kfs*8 | 杂合 | - | 否 |
| 家系2 | SALL4 | NM_020436 | exon2 | c.2210delG | p.G737Vfs*23 | 杂合 | - | 新变异 |
应用Sanger测序对家系1的高危胎儿(Ⅲ:5)和家系2的2个高危胎儿(Ⅳ:3,Ⅳ:4)的SALL4基因相应位点进行检测,发现家系1的胎儿(Ⅲ:5)和家系2的胎儿(Ⅳ:4)分别携带c.844delC p.(Q282Kfs*8)杂合变异和c.2210delG p.(G737Vfs*23)杂合变异,为Okihiro综合征患胎,经遗传咨询后,胎儿父母选择终止妊娠。家系2 的胎儿(Ⅳ:3)未携带c.2210delG p.(G737Vfs*23)变异,胎儿家属选择继续妊娠,经自然分娩出生,留取脐带血进行SALL4基因诊断与产前诊断结果一致,电话随访6个月,发育正常。
目前对Okihiro综合征的认识并不全面,暂无临床诊断标准,其患者表型是高度可变并涉及多个系统,某些患者表现出一系列临床表型,而一些患者仅表现出部分临床表型。即使在同一个家庭中的患者之间表型也可能不同,可能存在被误认为两种不同的疾病表型。加上某些临床表型与Holt-Oram、acro-reno-eye等疾病症状重叠[7],而且围产期超声影像存在局限性,在围产期对Okihiro综合征的临床诊断尤为困难。本研究中2个家系的患者临床表现差异很大,尤其是家系1第Ⅲ代中患者均通过胎儿超声发现,特别是第一个患胎(Ⅲ:3)既存在上肢畸形,同时存在严重的心脏畸形,胎儿的父母均无相关表现,这可能是这个胎儿未做相关遗传学检查的原因之一。先证者(Ⅲ:4)在孕12周发现上肢畸形,依据先证者的基因诊断和结合这2次的不良孕产史,才得以完成家系其他成员的临床诊断。因此,基因诊断在辅助Okihiro综合征的临床诊断中显得尤为重要。Okihiro综合征很罕见,以往研究表明大多数病例是散发的[8, 9],约5%Okihiro综合征患者为家族性遗传发病[10],有关Okihiro综合征患者在围产期的临床表现的报道很少。本研究的2个家系均为家族性遗传,家系1的2个受累胎儿均出现桡骨缺失和姿势异常,家系1的受累胎儿Ⅲ:3存在心脏畸形(永存动脉干和室间隔缺损),家系1的受累胎儿Ⅲ:4由于孕周小暂未发现心内结构异常。家系2的受累胎儿Ⅳ:2则主要问题为盆腔异位肾,骨骼方面仅见右手大拇指呈背曲状态,超声心动图显示心内结构正常。这些结果提示在围产期检测到胎儿存在骨骼上肢畸形合并心脏畸形或肾脏畸形的情况,应特别关注家族成员的骨骼、眼睛、心脏、肾脏等方面是否存在异常,结合家族史和家系成员临床表现有助于更准确地临床诊断。
SALL4基因变异的主要突变类型是无义变异、移码变异或外显子缺失产生的SALL4截短蛋白[9],截至目前,80个不同的SALL4变异收录在HGMD数据库,几乎所有的变异会导致至少一个ZFM的破坏。本研究发现的2个SALL4基因变异c.844delC和c.2210delG经预测均产生破坏至少一个ZFM的截短蛋白。已有研究表明截短蛋白可能抑制野生型SALL4蛋白的表达,呈显性负效应,SALL4蛋白缺乏或产生非功能性ZFM的蛋白导致单倍体不足,从而引起Okihiro综合征的发生[11, 12]。目前研究没有发现基因型和表型之间存在明确关联,ZFM结构域丢失数量和表型严重情况不一定相符,SALL4基因整个缺失导致的患者表型与点变异导致的患者表型并无明显差异[4]。本研究中2个家系患者成员临床表现差异很大,很难明确基因型和表型的关系。
总之,本研究通过对2个Okihiro综合征家系进行基因变异检测,发现SALL4基因变异c.844delC和c.2210delG,明确2个家系的致病原因,其中c.2210delG为新变异,丰富SALL4基因变异数据库,同时受累胎儿的超声影像完善了Okihiro综合征围产期临床表现,并为3个高危胎儿提供产前诊断,有效降低生育患儿的风险。
白莹, 吴庆华, 李福祯, 等. 2个汉族Okihiro综合征家系的SALL4基因新移码变异[J]. 中华医学杂志, 2023, 103(26): 2006-2010. DOI: 10.3760/cma.j.cn112137-20221206-02577.
所有作者声明不存在利益冲突

























