
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
巴特综合征(BS)是一种罕见的常染色体隐性遗传性疾病,为肾单位髓襻升支粗段水盐重吸收障碍导致的原发性肾失盐疾病,其主要特点为低血钾、低氯性代谢性碱中毒、高肾素活性和高醛固酮血症。BS根据致病突变所在基因不同分为5型,分别由SLC12A1基因(600839,1型)、KCNJ1基因(600359,2型)、CLCNKB基因(602023, 3型)、BSND基因(606412,4a型)、CLCNKB基因和CLCNKA基因(602024)共同突变(4b型)或CASR基因(601199,5型)突变造成。其中,2型致病基因KCNJ1位于染色体11q24.3,但因剪切位点的变异共产生5种转录本,可编码形成3种产物,肾脏外髓质钾通道(ROMK)1、ROMK2、ROMK3,分别由391个、372个、389个氨基酸组成,这三种产物在肾脏中均有表达。ROMK在髓襻升支粗段至远端肾单位均有分布,为内向整流钾通道,主要负责管腔膜侧钾离子的分泌。现报道1例2型BS患者。
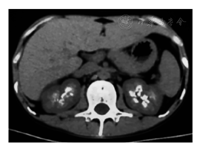
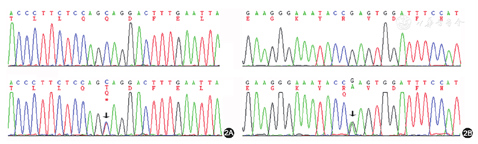
患者男,23岁。22岁时因多饮多尿1个月于2016年11月24日在我院肾内科就诊,门诊以尿崩症收入院。患者多饮多尿逐渐加重,尿量约10 L,比重1.002。每日饮水量约10 L,其中夜间饮水5 L。夜尿3~4次。患者无长期呕吐、腹泻史,且未服用利尿剂等药物。患者系足月顺产,父母非近亲结婚,患者其他直系亲属(父母及姐)均无相似病史。体检:血压117/79 mmHg(1 mmHg=0.133 kPa),身高172 cm,体重40 kg,BMI 13.52 kg/m2。实验室检查:血pH值7.46,血钾2.33 mmol/L,血钠137.00 mmol/L,血氯92.00 mmol/L,血二氧化碳结合力31.38 mmol/L,血钙2.54 mmol/L。尿钾19.32 mmol/L,尿钠51.00 mmol/L,尿氯42.00 mmol/L,尿钙1.33 mmol/L,尿镁1.47 mmol/L。尿肌酐1 777.00 μmol/L。尿钾排泄分数20%,尿钠排泄分数1.2%,尿氯排泄分数1.5%,尿钙/尿肌酐(mol/mol)0.75,尿镁排泄分数8.6%。血肌酐51.00 μmol/L,尿素氮7.59 mmol/L。25羟维生素D3 11.20 ng/ml。甲状旁腺素55.53 pg/ml。肾素-血管紧张素-醛固酮系统检测,卧位:肾素活性11.48 ng·ml-1·h-1,醛固酮192.07 pg/ml;立位:肾素活性>17.76 ng·ml-1·h-1,醛固酮695.76 pg/ml。查抗核抗体、抗可提取核抗原(ENA)谱均阴性。尿常规:尿pH值6.5, 尿比重<1.005,尿隐血1+,尿白细胞1+。超声检查:右肝前叶见强回声光斑0.6 cm×0.5 cm,边界清,双肾窦内多发高回声团,肾窦呈铸形,右肾较大1.9 cm×1.5 cm,左肾较大2.4 cm×2.2 cm。双肾CT示双肾钙质沉积及双肾结石(图1)。双侧99Tcm-MIBI(甲氧基异丁基异腈)甲状旁腺显像未见异常。禁水-加压素试验阴性。患者入院时表现为高血钙、肝右叶钙化灶、双肾结石、双肾钙质沉积,但甲状旁腺素无升高,双侧99Tcm-MIBI甲状旁腺显像未见异常,且复查血钙浓度2.4 mmol/L,故排除甲状旁腺机能亢进症。禁水-加压素试验阴性,排除中枢性尿崩。但患者低血钾、低血氯与原发性肾性尿崩(伴性遗传性肾性尿崩和常染色体隐性或显性肾性尿崩)不符,且患者临床特征与BS相符,故疑诊BS。查纯音听阈测定示双耳听力正常,排除4型BS。静脉+口服氯化钾治疗纠正血钾3.43 mmol/L,血钠144 mmol/L, 血氯102 mmol/L,血二氧化碳结合力30.67 mmol/L,于2016年12月7日出院,未行特殊治疗。2016年12月27日,患者及其家属抽取血样送往青岛金域医学研究所进行第二代高通量测序,对BS和Gitelman综合征(因Gitelman综合征往往与BS存在表型交叉)共7个相关基因(SLC12A1、KCNJ1、CLCNKB、CLCNKA、BSND、CASR、SLC12A3)进行测序,发现KCNJ1基因的2个新的杂合突变,确诊2型BS。一是位于5号外显子的无义突变c.865C>T,突变效应为p.Gln289*,该突变遗传自母亲;二是位于5号外显子的错义突变c.1013G>A,突变效应为p.Arg338Gln,该突变遗传自父亲。经检索人类基因突变库和最新文献,这2个突变是新发现突变。Sanger验证结果见图2。给予患者吲哚美辛0.31 mg·kg-1·d-1,多饮多尿症状当天即明显改善,尿量减少至2 000 ml/d。1周后复查血pH值7.43,血钾3.63 mmol/L, 血钠143.60 mmol/L,血氯103.51 mmol/L,血二氧化碳结合力29.25 mmol/L。随访至今,患者多饮多尿症状无反复,多次复查血酸碱度和电解质指标均正常。
自1996年发现ROMK的编码基因突变可导致出生前BS以来,世界范围内报道的KCNJ1基因突变已达68种,包括2个插入突变,5个无义突变,7个缺失突变,54个错义突变。
本例患者22岁发病,以多饮多尿1个月为主诉入院,初步诊断为尿崩症。后考虑其多饮多尿的症状和低血钾、低氯性代谢性碱中毒、高血浆肾素活性、高醛固酮血症、尿钾钠氯排泄分数升高的生化特点及正常血压符合BS的表现,故疑诊为BS。患者系足月顺产,儿童期亦无BS症状,属成年后发病,符合经典型BS。但肾钙质沉积和肾结石多见于出生前BS,在经典型BS患者中较为少见。而且本例患者已排除假性及继发性BS的可能,故有必要进行相关致病基因检测以明确诊断。
通过第二代高通量测序,检测到KCNJ1基因5号外显子的复合杂合突变:c.865C>T,p.Gln289*和c.1013G>A,p.Arg338Gln。本例患者基因确诊为2型BS,并排除其他型BS和Gitelman综合征。无义突变p.Gln289*导致翻译提前终止于第289位密码子而形成截短的肽链。错义突变p.Arg338Gln位于ROMK羧基端,经Grantham Matrix评分系统预测存在中度致病可能,MutationTaster和Polyphen2预测其存在致病可能,SIFT预测为良性突变。另外,经同源序列分析,KCNJ1基因编码蛋白ROMK的338位氨基酸在8种不同生物中高度保守,但在人类ROMK家族中,第338位氨基酸并不保守。
经检索文献,晚发2型BS亦有报道。Huang等[1]于2014年报道了1例以腰部疼痛2个月为主诉入院的35岁男性患者,KCNJ1基因检测发现纯合突变c.658C>T(p.Leu220Phe),由此作者提出2型BS表型严重程度存在较大异质性。Sharma和Linshaw[2]于2011年报道了1例足月顺产,幼儿期才逐渐出现多饮多尿的13岁女童,KCNJ1基因检测为复合杂合突变c.268G>T(p.Gly90Trp)和c.632T>G(p.Ile211Ser),作者推测患者症状晚发的原因可能是携带以上两种错义突变的产物仍残存部分活性。
本例新发现的错义突变c.1013G>A(p.Arg338Gln)位于ROMK的羧基端,而羧基端已被证实在ROMK的孔隙特性中起决定性作用。Cho和Guay-Woodford[3]曾报道1例携带KCNJ1基因p.Arg338*和 p.Met357Thr复合杂合突变的2型BS患者,该患者的母亲有孕期羊水过多和早产史,存在双肾钙质沉积。但其病情演变良性化发展,低血钾、低氯性代谢性碱中毒、高肾素活性和高醛固酮血症等生化表现轻微,且经吲哚美辛治疗后,血钾、二氧化碳结合力和尿钙/尿肌酐均可达正常水平,提示该患者的临床表现较其他2型BS患者偏轻。作者推测其相对较轻的临床表型可能与p.Met357Thr仍残存部分功能有关。在人类基因突变数据库中,检索到与p.Arg338Gln相邻且同处于羧基端的4个突变位点p.Leu320Pro、p.Phe325Cys、p.Met357Thr、p.Leu359Arg,并对其进行同源序列分析,结果显示,325位和338位氨基酸高度保守。至目前为止,所报道的距离羧基端最近的突变是p.Leu359Arg,有研究发现携带此突变的正常人血压偏低,提示该突变可能通过轻度的利钠作用对高血压发病有保护作用[4]。本例患者发病年龄较晚,临床表现较轻,推测可能与新发现的错义突变p.Arg338Gln致病性较弱、仍残留部分功能有关,但需进一步体外功能研究证实。
本例患者应用吲哚美辛和氯化钾联合治疗效果显著,但疾病演变和肾功能仍需长时间观察和随访。

























