
报道1例以横纹肌溶解、急性肾衰竭、急性肝衰竭起病的病例,最终诊断为多酰基辅酶A脱氢酶缺乏症,加用大剂量维生素B2后病情一度稳定,终因合并肺部感染、呼吸衰竭、心力衰竭而死亡。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者男,22岁。因"肌痛、乏力1个月,加重伴尿少、意识改变3 d"于2016年12月23日收入我院MICU病房。患者为在校大学生,2016年12月初活动量增大(跑步600 m)后出现肌肉酸痛、乏力,以双下肢近端肌肉为著,同时出现纳差厌油腻。12月20日患者四肢无法抬离床面,并出现浓茶色尿、尿量明显减少,伴淡漠、懒言,宿舍同学将其送至当地医院急诊。查血常规:白细胞25.23×109/L,血红蛋白浓度147 g/L,血小板计数156×109/L;肝肾功能:丙氨酸转氨酶(ALT)1 020 U/L,总胆红素171.6 μmol/L,直接胆红素43.4 μmol/L,肌酐175 μmol/L。予床旁血液滤过治疗,效果不佳。12月22日转至我院急诊,入室时意识障碍,呼之不应,体温38 ℃,血压80/40 mmHg(1 mmHg=0.133 kPa),心率63次/min,血气分析示代谢性酸中毒(储氧面罩):pH 7.252,二氧化碳分压43.9 mmHg,氧分压239 mmHg,碳酸氢根18.7 mmol/L,乳酸3.9 mmol/L。肝肾功能进一步恶化:ALT 1 374 U/L,总胆红素87.6 μmol/L,直接胆红素72.9 μmol/L,血氨252.0 μmol/L,血钾7.1 mmol/L,尿素31.95 mmol/L,肌酐462 μmol/L。凝血酶原时间19.0 s,活化部分凝血活酶时间大于150 s。同时发现肌酶显著增高:肌酸激酶大于100 000 U/L,肌酸激酶同工酶MB大于1 200 μg/L,心肌肌钙蛋白I 0.770 μg/L,肌红蛋白46 518 μg/L。考虑"横纹肌溶解,急性肾衰竭,急性肝衰竭"。予支持治疗,并给予降钾、保肝、白醋灌肠及床旁血液滤过治疗。次日患者意识好转,血压130/90 mmHg,心率90次/min,四肢肌力、肌张力可,心肺腹体检无特殊发现,复查血钾4.4 mmol/L,转入MICU病房。入科后予间断血液净化治疗,患者体温恢复正常,仍持续无尿,反复高钾、低血糖。复查血生化:总胆红素从80.9 μmol/L➝104.8 μmol/L,直接胆红素从72.7 μmol/L➝97.5 μmol/L,肌酸激酶从>105 U/L➝1 136 U/L,肌酸激酶同工酶MB从>1 200 μg/L➝28.4 μg/L,肌红蛋白从46 518 μg/L➝4 869 μg/L。完善病因筛查,感染方面:嗜肝病毒检查均(-);免疫方面:抗核抗体谱18项显示,抗PM-Scl弱(+),余(-),肌炎抗体谱(-),自身免疫性肝炎相关抗体谱均(-);毒物方面:血液中铅、汞、铊、铬、镉、砷无升高,血、尿中未检测到明确毒物;代谢方面:糖化血红蛋白、甲状腺功能均正常,总胆固醇5.02 mmol/L,总甘油三酯4.29 mmol/L,胸腹CT示重度脂肪肝。
马杰医师(肾内科)横纹肌溶解根据起病诱因可分为3类:创伤性、非创伤劳累性与非创伤非劳累性。本例有加强运动的诱因,需考虑非创伤劳累性横纹肌溶解。非创伤劳累性横纹肌溶解见于未经训练的个体过度运动、热射病或代谢性肌病。用单纯过度运动诱发横纹肌溶解来解释本例病情存在以下困难:(1)急性肝衰竭:横纹肌溶解患者中约25%合并肝损伤,多见于有基础肝病的患者,表现为肝酶一过性升高,一般不会出现严重肝衰竭。患者发现高脂血症、重度脂肪肝,应考虑肝脏病变是否与横纹肌溶解为两个独立疾病,或某种共同诱因同时导致了横纹肌溶解与急性肝衰竭。(2)疗效不佳:横纹肌溶解诱发急性肾衰竭经积极支持治疗,80%的患者可在1~2周内进入多尿期,最终肾功能可恢复至正常。本例积极治疗已有10 d,仍处于无尿状态,需考虑某种持续存在的病因仍未去除。另一类引起非创伤劳累性横纹肌溶解的少见病因为代谢性肌病,可伴肝脏病变,在过度劳累、饥饿等条件下诱发急性代谢危象,表现为横纹肌溶解、急性肝衰竭等。本例需警惕此类疾病,应进一步追问童年病史,需完善肌肉活检等进一步明确。
江伟医师(MICU)横纹肌溶解的治疗原则为去除诱因,支持治疗。对于肾功能轻度受损的患者,需保证入量,积极水化、碱化,预防并纠正高钾、高尿酸血症。对于急性肾衰竭的患者,出现急诊透析指征时,需开始透析治疗。透析方式首选血液透析、高剂量持续肾脏替代治疗(CRRT),血液透析对于电解质紊乱纠正作用更强,CRRT则对容量管理更有优势。本例患者在行CRRT过程中仍有反复高钾,后转入肾内科病房开始高频率血液透析后,血钾水平可基本维持正常。腹膜透析因其充分性欠佳,不推荐应用于横纹肌溶解合并急性肾衰竭的患者中。
本例患者为青年男性,急性起病,临床表现为横纹肌溶解伴急性肾衰竭、急性肝衰竭。经MICU积极支持治疗,生命体征虽已趋平稳,但持续无尿、高钾以及反复低血糖,提示患者的肌肉破坏、肝损害仍在继续,下一步目标在于尽快明确诊断,对因治疗。
患者于12月30日转入肾内科病房。追问病史,患者自幼活动量较同龄人差,剧烈活动后易乏力,父母否认近亲结婚,有1弟,14岁,体健。结合患者病史,考虑代谢性肌病可能性大,建议行肌肉活检、肝穿刺活检,但患者家属因经济困难拒绝行进一步活检,同意留取血、尿送检有机酸检测。2017年1月5日血尿有机酸检测回报:氨基酸血症,有机酸血症(乳酸、酮体、戊二酸和2羟基戊二酸明显增高),脂肪酸代谢缺陷(C4,C5,C6,C8,C10,C12:1,C14:2,C5DC,C4/C3,C5/C4,C8/C2,C8/C3,C8/C16,C14:1/C16增高;C0,C0/C16,C3/C16降低*)。结果回报符合多酰基辅酶A脱氢酶缺乏症(MADD),伴继发性肉碱缺乏。立即加用维生素B2 100 mg 3次/d口服、左旋肉碱2 g 4次/d静点,调整肠内营养为高碳水化合物、低脂、低蛋白饮食,并加用中链甘油三酯10 g 3次/d口服。此后,患者病情开始出现稳定并好转的趋势,尿量逐渐升至120 ml/d,透析频率由1次/d延长至1次/2 d,未再出现高钾。肝脏方面,胆红素升高速度减缓并出现回落趋势,总胆红素从104 μmol/L➝407 μmol/L➝361 μmol/L,直接胆红素从97 μmol/L➝376 μmol/L➝343 μmol/L,凝血、血氨、乳酸恢复正常,未再出现低血糖(图1)。
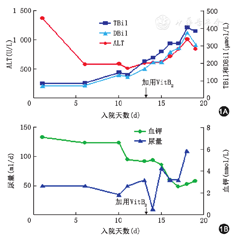
注:ALT为丙氨酸转氨酶;TBil为总胆红素;DBil为直接胆红素
马杰医师(肾内科)代谢性肌病是一组遗传代谢性疾病,是非创伤劳累性横纹肌溶解的罕见病因。其中MADD是一种罕见的常染色体隐性遗传病,为电子转运黄素蛋白(ETFA、ETFB)或电子转运黄素蛋白脱氢酶(ETFDH)缺陷,与核黄素亲和力下降,多种脂肪酸、支链氨基酸代谢异常,引起肝脏和肌肉病变。早发型患者常于新生儿期、婴幼儿期死亡。迟发型平均发病年龄19岁,多表现为慢性肌肉耐力下降,可伴高脂血症、脂肪肝,约1/3患者在感染、劳累、饥饿等诱因下可出现急性代谢失代偿,表现为酸中毒、横纹肌溶解、急性肾衰竭和急性肝衰竭[1]。在急性发作期出现典型的血尿有机酸代谢谱(各种脂酰肉碱、乳酸、酮体、戊二酸和2羟基戊二酸明显增高,C0明显降低*),但是在无症状期可呈假阴性。治疗方面,推荐高碳水化合物、低蛋白、低脂饮食,大部分迟发型患者对大剂量维生素B2(即核黄素,推荐剂量5~10 mg·kg-1·d-1)反应好,推测增加核黄素底物水平可能一定程度代偿ETF/ETFDH与核黄素的结合困难[2]。亦有文献报道补充左旋肉碱、辅酶Q10可能获益。然而,疗效研究仅在一般情况良好的患者中进行,对已出现严重代谢失衡的急性患者治疗方面的经验十分有限,有个例报道虽加用大剂量维生素B2治疗,病情一过性好转随即加重死亡[3]。
本例患者临床高度怀疑MADD可能,病情危重、进展快、经济条件困难,无法行肌肉活检等待病理结果,于是我们直接进行了血尿有机酸代谢学检测,结果符合MADD表现,遂加用大剂量维生素B2和左旋肉碱治疗,患者肝肾功能及代谢情况逐步稳定并有好转趋势,进一步印证了本病的诊断,亦对此类急性暴发型患者的治疗有所提示。下一步应争取行基因检测,以明确诊断并精确分型。
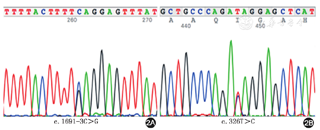
司锘研究员(遗传学组)患者ETFA、ETFB及ETFDH基因测序结果回报:ETFDH基因存在复合杂合突变,包括1个已知的致病突变(c.1691-3C>G)[4]与1个未报道的错义突变(c.326T>C,pI109T)(图2)。其中错义突变(c.326T>C,pI109T)虽尚未报道是否致病,但是千人数据库(1000 Genomes Project)、Ensembl数据库均未发现正常人群存在该突变。Polyphen-2、SIFT软件对pI109T突变进行预测,结果均提示该位点氨基酸序列在多个物种中进化高度保守,且氨基酸突变对蛋白质功能造成致病性影响可能性大。而且该氨基酸所在区域为ETFDH蛋白最重要的功能区域——核黄素结合区(FAD),目前已知的大多数MADD致病突变均位于该区域。这些证据提示本例发现的ETFDH基因突变(c.326T>C,pI109T)很可能是1个尚未被报道的致病突变。
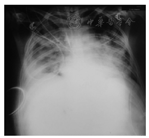
患者病情趋于稳定,2017年1月9日突发憋气,双肺新发满布哮鸣音、多发湿啰音,床旁胸部X线片提示急性肺水肿(图3),予床旁血液滤过(超滤量4 L)、无创通气,呼吸困难可好转。2017年1月10日出现发热,体温38.7 ℃,伴氧合下降,动脉血气示呼吸衰竭(pH 7.319,二氧化碳分压49.4 mmHg,氧分压53.7 mmHg,碳酸氢根24.6 mmol/L,乳酸0.9 mmol/L),考虑呼吸肌受累不除外,合并肺部感染,此后氧合迅速下降,难以维持,患者家属拒绝有创抢救,患者于2017年1月10日死亡。
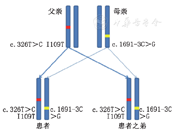
司锘研究员(遗传学组)我们对患者父母及弟弟进行了ETFDH基因测序,结果提示患者弟弟ETFDH基因存在与患者相同的复合杂合突变(图4)。通知患者父母,并对患者弟弟进行了血、尿有机酸检测,结果提示其存在复合型脂肪酸氧化异常伴可疑的继发性肉碱缺乏,虽非特异却符合MADD的代谢学异常(C8,C10,C8/C2,C8/C3,C8/C12,C8/C16增高,C0降低)。考虑患者弟弟MADD诊断可能性大,遂给予其大剂量维生素B2、左旋肉碱,嘱其避免过度劳累、饥饿、剧烈运动。
李雪梅教授(肾内科)MADD目前已知的致病基因包括3个:ETFA、ETFB、ETFDH,中国患者93%为ETFDH基因突变,基因型与表现型的相关性目前尚不清楚[5,6]。本例患者病情渐趋稳定过程中突发急性心力衰竭、呼吸衰竭,病情迅速恶化死亡。不除外心肌受累、呼吸肌受累合并肺部感染可能。MADD早发型可伴心肌、呼吸肌受累,迟发型仅有个案报道[3,7,8]。本例患者基因型为复合杂合突变,其中1个位点突变尚未见报道,表现型差异不除外与基因型相关。
大部分迟发型MADD患者对大剂量维生素B2反应良好,但在急重症中治疗经验非常有限,本例加用大剂量维生素B2后病情一度趋于稳定,但终因合并肺部感染,病情急剧加重而死亡。这也提示该病贵在早期诊断、早期治疗。本例患者的弟弟具有与患者相同的复合杂合突变,目前尚未出现典型的临床症状,考虑可能的原因如下:(1)年龄尚小。我国的研究发现,MADD患者平均发病年龄约为19.2岁[1],而患者弟弟年仅16岁,不除外与年龄尚小尚未表现出特异的临床表现相关。(2)MADD临床异质性大。已有的家系报道均发现,该病即使在携带相同致病突变的家系成员中,仍有极大的临床异质性。曾有文献报道,姐妹2人携带相同的致病突变,妹妹反复出现急性代谢危象,而姐姐仅表现为轻微的运动耐量下降和血、尿有机酸代谢谱异常[3]。值得注意的是,患者弟弟的血、尿有机酸代谢产物检测已提示其存在脂肪酸代谢异常,虽未见特异性MADD改变,但其多种长度的脂肪酸增高、C0减低均符合MADD表现,部分MADD患者仅在病情急性加重时出现典型的血、尿有机酸谱表现,而在急性发作间期可出现假阴性。对于MADD患者,推荐一旦基因诊断确立,即开始补充维生素B2,并注意避免过度劳累、饥饿、剧烈运动。已嘱患者弟弟加用维生素B2 100 mg 3次/d口服及左旋肉碱口服,门诊随诊,如及适婚年龄,需进行该病的遗传咨询。
最终诊断:多酰基辅酶A脱氢酶缺乏症(迟发型),横纹肌溶解,急性肾损伤,呼吸肌受累可能,呼吸衰竭,肺部感染,心肌受累不除外,急性心功能衰竭,脂肪肝(重度),急性肝衰竭。
本例患者以横纹肌溶解、急性肾衰竭、急性肝衰竭起病,病情危重、进展快,虽诊断为MADD,最终因病情加重去世。回顾诊治过程,本病贵在早期诊断、尽快治疗,体会如下:(1)代谢性肌病是横纹肌溶解的少见病因,对于年轻、长期活动耐量较差、合并肝功能严重受损的患者,特别是积极支持治疗后仍有肌肉持续破坏、肝功能持续恶化者,更需高度警惕本病,积极完善肌活检、血尿有机酸检查明确诊断。(2)回顾文献发现,对于本病急重症患者的病例报道少见,且缺乏在这些患者中的治疗经验,一方面本病多以慢性轻度活动耐量下降为主要表现,急重症患者相对少见,另一方面临床医生对于本病了解仍较少,早期诊断困难,而患者病情进展快,难以坚持到诊断明确。本例报道以横纹肌溶解、急性肾衰竭、急性肝衰竭起病的MADD患者,并分享我院在该患者治疗中的一点有限经验和教训,希望对临床医生有所帮助。(3)本病为常染色体隐性遗传病,推荐一旦基因诊断确立即开始补充维生素B2,延缓病情发展并预防病情急性加重。因此,对于明确诊为MADD患者的一级亲属推荐进行致病基因位点测序,以助于发现未急性发病的患者,及早开始干预。(4)本例患者基因检测新发现一高度可疑致病的错义突变(c.326T>C,pI109T),需要进一步的功能学研究明确该突变的致病意义及基因型-临床型之间的关系。
*注:C4即指含有4个碳素的脂肪酸肉碱(丁酰肉碱),C5为含有5个碳素的脂肪酸肉碱(戊酰肉碱),其余同理。C0指游离肉碱。C12:1指含有1个双键的12碳素脂肪酸肉碱(十二碳烯酸肉碱)。同理,C14:1为十四碳烯酸肉碱,C14:2为十四碳二烯酸肉碱。C5DC为戊二酰肉碱。MADD患者线粒体呼吸链多种脱氢酶功能受阻,因此短链、中链及长链的酰基肉碱水平均升高,且继发肉碱缺乏(C0降低),与本例吻合。

























