
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
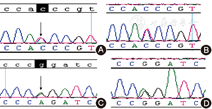
女,因"新生儿筛查发现半乳糖(galactose)(包括半乳糖和1-磷酸半乳糖)增高"召回。出生第3天足跟采血行半乳糖血症(galactosemia,GAL)筛查,血半乳糖浓度122 mg/L(正常<100 mg/L),生后7 d召回复查时无主诉不适;半乳糖215 mg/L,体格检查正常;血生化检测发现谷氨酰转肽酶61 U/L(正常5~50 U/L)、总胆汁酸17 μmol/L(正常0~12 μmol/L)轻度增高;眼科检查及腹部超声检测未见异常。第1胎第1产,孕38周自然分娩,出生体重3 kg,Apgar评分10分。在家长知情同意后抽外周静脉血送深圳华大基因公司检测GAL基因,送检尿苷二磷酸葡萄糖4表异构酶(GALE)、半乳糖激酶及半乳糖1磷酸尿苷转移酶(GALT)。发现GALE基因存在c.505C>T (p.R169W)和c.452G>A (p.G151D) 2个错义突变位点(图1),父母外周静脉血Sanger验证发现母亲携带c.505C>T ,父亲携带c.452G>A (图1)。红细胞GALE酶活性检测为正常对照的42%。GALT酶活性未见异常。
给予无乳糖奶粉每天60 g,不足部分母乳补充,每间隔1个月来本中心随访。第2个月生化指标全部恢复正常。监测至4个月,半乳糖波动于202~251 mg/L,改完全无乳糖奶粉喂养,第5个月后半乳糖恢复至正常范围。第6个月、12个月、24个月身高、体重、头围检测均正常。第9个月眼裂隙灯检查正常。12月龄时应用Bayley发育量表检测语言发育指数(120)、运动发育指数(125)均正常。
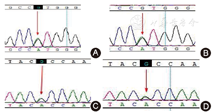
男,因"新生儿筛查发现半乳糖增高"召回。出生第3天足跟采血行GAL筛查,半乳糖浓度255 mg/L,生后10 d召回复查时无不适主诉,半乳糖353 mg/L,体格检查正常;血生化检测发现血总胆红素29.3 μmol/L(正常0.0~19.0 μmol/L)、直接胆红素21.0 μmol/L(正常0.0~10.0 μmol/L)、谷草转氨酶59 U/L(正常5~55 U/L)、总胆汁酸202 μmol/L增高;眼科检查及腹部超声检测未见异常。患儿为第2胎第1产,母孕40周自然分娩,患儿出生体重3.5 kg,Apgar评分10分。在家长知情同意后抽外周静脉血送深圳华大基因公司检测GAL基因,发现GALE基因存在c.280G>A (p.V94M)和c.925G>A (p.A309T) 2个错义突变位点(图2),父母外周静脉血Sanger验证发现母亲携带c.280G>A ,父亲携带c.925G>A (图2)。红细胞GALE酶活性检测为正常对照的38%。GALT酶活性未见异常。
完全无乳糖奶粉喂养,每间隔1个月来本中心随访。第2个月生化指标全部恢复正常。目前随访至17月龄,眼裂隙灯检查未见异常;身高、体重、头围检测均正常;能翻身,会认人。
2例父母均为汉族,体健,非近亲结婚,无家族遗传病史。
GAL是某种酶缺乏致半乳糖利用障碍引起一系列症状的常染色体隐性遗传病。目前发现有3种酶缺陷均可致该病,包括半乳糖激酶、GALT和GALE。GALE位于半乳糖代谢的第3步,缺陷导致半乳糖血症Ⅲ型(OMIM 230350) (UDP-galactose-4'-epimerase deficiency, GALED)[1]。由于GALED较罕见,该型的群体发病率、基因型和表现型特点、酶活性与基因型的关系及长期预后并不完全清楚[2,3]。国内尚无明确基因诊断的GALED病例相关报道。
GALED发病率具有明显的种族差异。非洲裔美国婴儿和日本婴儿中发病率分别约1∶10 000、1∶250 000[1],而欧洲裔美国婴儿约1∶70 000[4]。中国深圳报道GAL总发病率约1∶50 000[5]。本中心筛查新生儿599 140例,经基因检测确诊GALED 2例,检出约1∶300 000,提示GALED在浙江省较罕见,与日本发病率接近。
GALED临床表现具有高度异质性。依据GALE酶活性在不同组织细胞的改变,临床分为全身型、周围型、中间型三型[6,7,8]。全身型患者临床症状重,表现类似GALT缺陷患儿,需早诊断早治疗[9];而周围型和中间型预后好[3]。周围型通常不需限制半乳糖饮食,中间型治疗方案尚未明确,有报道未经治疗的中间型GALED在儿童早期出现运动及认知发育落后[8]。但由于国内临床病例组织标本较难获得,酶活性检测尚未普及,该分型方法较难在临床开展。目前多建议婴儿周围型病例不需饮食治疗,但需长期随访;中间型及重症型病例需无乳糖奶粉治疗及长期随访。年长儿虽对半乳糖耐受性增加,中间型及重症者仍需持续无乳糖奶粉喂养并长期随访[10]。由于新生儿筛查发现病例多无临床症状,立即区分轻重症较困难,建议先饮食治疗,明确为轻症型可停无乳糖奶粉。本病长期预后主要取决于疾病类型及诊治早晚:轻症型,预后良好;中间型、重症型即便经饮食治疗仍可能出现认知障碍、发育落后等长期并发症[6,10]。
人类GALE基因位于染色体1p36,由11个外显子组成[11]。目前已报道GALED有23个突变位点,包括22个错义突变和1个无义突变(http://www.hgmd.cf.ac.uk/ac/all.php)。由于报道病例较少,GALED基因型-表现型关系还不十分清楚。一般认为临床表型取决于各基因决定的相关酶活性的大小[8];复合杂合突变病例临床表型取决于残余酶活性较高的那个基因,如GALE重症型基因G90E复合轻症型基因病例时临床表现为周围型[11]。本组2例均为复合杂合突变,其中例1的R169W位点在韩国人群较常见,其酶活性为正常对照的60%,复合杂合突变时临床表现为周围型[12,13]。G151D位点未见文献报道,经SIFT、PolyPhen-2蛋白功能预测为有害,同时经多重比对序列分析,该位置上的氨基酸较为保守,根据PDB蛋白结构数据库(http://www.rcsb.org/pdb/protein/Q14376),G151D在GALE序列中位于一段松散的无规卷曲,该区域暂不清楚有何功能或作用,但非常靠近已报道的致病突变T150M,T150M酶活性为正常对照的24.72%,临床表现为中间型[13]。例1 GALE红细胞酶活性42%,召回时仅见半乳糖及其肝功能轻度改变,予无乳糖奶粉治疗后随访至12月龄,体格发育及Bayley发育评分均正常,未见白内障改变,符合轻症型GALED表现。例2中V94M突变是GALED已知重症型致病突变,酶活性为正常对照的3.06%[13]。而A309T未见文献报道。该突变是错义突变,导致编码的氨基酸由非极性的丙氨酸变为极性的苏氨酸,氨基酸性质发生改变;在dbSNP、1000G、1000G ASN数据库中的频率全部为0;经SIFT、PolyPhen-2蛋白功能预测为有害。鉴于例2 GALE酶活性较高(38%),在等位基因V94M为已知重症型基因情况下临床随访过程中未发现异常表现,提示A309T可能是GALED轻症型基因。但由于随访时间较短,需进一步验证。
血半乳糖增高还可见于希特林蛋白缺乏症,葡萄糖/半乳糖吸收不良症等,临床应注意鉴别。
由于GALED重症型临床表现危重,2例患儿虽经早期无乳糖奶粉治疗预后良好,但新生儿召回时已有肝功能损害,治疗前半乳糖呈上升趋势,建议GALED患儿父母若再次妊娠时行产前诊断。

























