
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
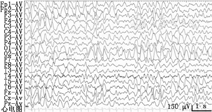
患儿女,4岁。因"间断抽搐2年余"于2016年2月就诊于北京大学第一医院。患儿足月自然生产,生后无窒息史。13月龄无明显诱因首次出现发作,表现为双眼左斜,左侧肢体抽搐,牙关紧闭、面色青紫,持续约5 min,24 h内发作3次,未予治疗。间隔7个月后出现第2次发作,仍为24 h内发作3次,表现同前,予托吡酯治疗。2岁2月龄时再次出现发作,表现为头、眼向一侧偏斜,不伴肢体抽搐,予托吡酯加量后发作控制不佳。后发作逐渐频繁,平均1~2个月发作一簇,于1~2 d内频繁发作,最多时1 d可发作7~8次,多表现为一侧身体抽搐,偶尔出现四肢抽搐,应用托吡酯、丙戊酸、硝西泮联合治疗,发作频率、程度均无明显改善。期间行脑电图检查示前头部、后头部及广泛性游走性慢波节律(图1)。确诊前末次发作为3岁10月龄时,24 h内发作7~8次。血液生化、氨基酸,尿液有机酸筛查等均未见异常。磁共振成像(MRI)检查示双侧侧脑室后角旁脑白质轻度髓鞘化不全,额部脑沟轻度增宽。患儿1岁6月龄会走路,至4岁时仍走路不稳、易跌倒;语言表达能力差,仅能说简单词语;精神较差,双眼无神,交流少。Gesell发育评估(4岁时)示轻-中度发育落后。此次于我院疑诊为限于女性的癫痫伴智力低下(EFMR)并行癫痫相关基因包检测,意外检出ALDH7A1基因复合杂合突变(NM_001182.4):c.1279G>C(p.Glu427Gln)和c.986G>A(p.Arg329Lys)(图2),分别遗传自父母,c.1279G>C为已报道致病性突变,c.986G>A未见国际报道,经保守性及致病性分析认为可致病,确诊为吡哆醇依赖性癫痫(PDE)。确诊后预计添加大剂量维生素B6,治疗当天出现1次发作,表现为双眼右斜,持续2 min,给予维生素B6 180 mg/d分3次口服后发作控制,未出现既往均会出现的成簇发作。维生素B6维持治疗1周后逐渐减停抗癫痫药。添加维生素B6治疗前及治疗2个月后的尿液哌啶酸浓度分别为2.09、1.64 mmol/mol肌酐(正常范围:0.12~3.52 mmol/mol肌酐)[1]。随访6个月,目前仅口服维生素B6 180 mg/d,未再出现癫痫发作。家长诉患儿精神状态较前明显好转,反应较前增快,记忆力明显增强;走路变稳,很少跌倒,可以跑;说话较前明显清晰,可以简单沟通。
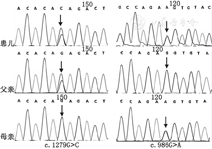
箭头示突变位点
PDE典型临床表现为新生儿期或婴儿早期即出现难治性癫痫发作,发作形式多样,抗癫痫药不能控制发作,而高剂量吡哆醇有效,需终生维持治疗,一旦停用会在1~51 d内复发[2]。然而,约1/3的患儿临床表现不典型,包括起病晚,可晚至3岁[3];发作最初可通过抗癫痫药控制或最初应用吡哆醇无效等[4]。此患儿为局灶性发作,多种抗癫痫药无效,最终被大剂量维生素B6控制,符合典型临床表现。但其起病较晚,13月龄首次出现发作;发作有明显丛集性特点,多在1~2 d内频繁发作,最长可间隔7个月不发作,且多数发作仅表现为一侧肢体抽搐,均为较少见的不典型临床表现,曾疑诊为EFMR。有报道停用维生素B6后最长可5.5个月无发作[5],而此患儿在未用任何药物治疗的情况下最长可7个月不发作。Coulter-Mackie等[6]报道E427G、W203G、N301I和T325R等突变可能与晚发型PDE有关,但PDE的基因型-表型关系尚不明确,相同的基因突变引起的临床表现存在一定的差异,可能受其他因素的影响,如饮食中赖氨酸、吡哆醇的摄入量及合成、分解代谢的个体差异等[6]。PDE患儿体内α-氨基己二酸半醛、Δ1-四氢吡啶-6-羧酸及哌啶酸浓度可升高。尿液哌啶酸浓度在治疗之前可高至正常高限的17倍,长期应用吡哆醇治疗后可明显降低甚至恢复正常[1]。本例患儿应用维生素B6治疗前、后尿液哌啶酸浓度均正常,可能与起病年龄相对较晚且检测时年龄相对较大,而尿液哌啶酸在年龄较大的儿童中分泌不显著[3]有关;同时可能与其基因型有关[7]。原因不明的难治性癫痫均应考虑PDE的可能性,并尽早试用维生素B6治疗,有反应者进一步行基因检测明确诊断,避免漏诊或误诊。

























