
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿男13岁。主因反复口腔白膜、皮疹伴呼吸道感染13年,生长发育迟缓12年,反复血细胞减少6年,加重2个月入院。患儿生后2个月开始反复出现双侧颊黏膜白色膜状物,胸背部硬性结痂皮疹,支气管肺炎(多次痰培养结果为白色念珠菌)。考虑为慢性皮肤、黏膜念珠菌病,呼吸道真菌感染。抗真菌治疗有效。患儿现肺部仍残留肺脓肿及支气管扩张。患儿1岁6月龄发现身材矮小,至今患儿身高、体重仍低于同龄儿童正常身高、体重值第三百分位。甲状腺功能示三碘甲腺原氨酸、甲状腺素降低,促甲状腺激素(TSH)升高,外源性甲状腺素补充治疗有效。7岁起反复出现血细胞减少,应用丙球蛋白和激素治疗后血小板可升至正常,但贫血无改善。入院前2个月血常规检查发现血红蛋白中度减低,网织红细胞增多,血小板中至重度减少,白细胞正常,未予诊治。患儿父母非近亲结婚,患儿系第6胎第6产,胞姐5人,均体健。患儿外祖父死于食管癌,余家族史无异常。
体格检查:患儿身高129 cm,体重20 kg(均位于同龄儿童正常身高、体重值第三百分位以下),重度营养不良,发育迟滞,精神反应欠佳。巩膜黄染,口腔黏膜白斑,皮肤苍白,可见全身皮肤散在瘀点、瘀斑,无皮肤及甲床真菌感染;听诊双肺呼吸音粗,心脏体检未见明显异常;肝肋下4 cm,肝区无压痛,脾肋下1 cm,质地中等,边缘钝。
辅助检查:白细胞(3.21~20.01)×109/L,白细胞分类中性粒细胞0.64~0.81,淋巴细胞绝对计数(0.52~1.90)×109/L,血红蛋白(55~92)g/L,血小板(10~357)×109/L,网织红细胞0.1~0.39;骨髓检查:骨髓增生明显活跃,粒系、巨核系增生活跃,红系增生旺盛,占0.6,以中幼红为主。住院期间3次抗人球蛋白试验均为阳性(++++),血管性血友病因子裂解蛋白酶活性0.7。尿常规未见异常。便潜血阳性。血生化检查:血钾2.73 mmol/L,钠129.6 mmol/L,直接胆红素1.38 μmol/L,间接胆红素6.65 μmol/L,乳酸脱氢酶496 U/L。免疫功能:免疫球蛋白(Ig)A 2.76 g/L,IgG 18.1 g/L,Ig M 0.963 g/L,Ig E 4.87 kU/L;白介素10: 22.23 ng/L,白介素6: 9.71 ng/L,白介素4: 0 ng/L;外周血淋巴细胞亚群:CD3-CD19+淋巴细胞0.25(正常0.05~0.18)、NK细胞0.009(正常0.070~0.400)、Treg细胞0.038(正常0.041~0.094)、Th17细胞0(正常0.002~0.024)。血液风疹病毒、巨细胞病毒、单纯疱疹病毒及弓形体抗体均阳性。乙肝五项、人类免疫缺陷病毒、丙型肝炎病毒、梅毒阴性。2次测定MLC+IFN-γA、B结果均为0。痰培养:白色念珠菌。
经父母知情同意后,采用一代测序的方法,对患儿及其父母血液行免疫缺陷基因筛查(送北京康旭医学检验所检)(表1):在受检者STAT1基因发现c.1154C>T,为自发的杂合核苷酸变异,该变异导致第385号氨基酸由苏氨酸变为甲硫氨酸(p.Thr385Met),为错义变异,呈常染色体显性遗传方式,可致慢性皮肤念珠菌感染;此外患儿ADAMTS13基因、STAT2基因及AIRE基因均发现杂合核苷酸变异,ADAMTS13杂合基因均来自父亲,AIRE、STAT2杂合基因源自母亲。
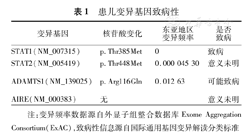
患儿变异基因致病性
患儿变异基因致病性
| 变异基因 | 核苷酸变化 | 东亚地区变异频率 | 是否致病 |
|---|---|---|---|
| STAT1(NM_007315) | p.Thr385Met | 0 | 致病 |
| STAT2(NM_005419) | p.Thr448Met | 0.000 045 30 | 意义未明 |
| ADAMTS1(NM_139025) | p.Arg116Gln | 0.012 63 | 可能致病 |
| AIRE(NM_000383) | 无 | 意义未明 |
注:变异频率数据源自外显子组整合数据库Exome Aggregation Consortium(ExAC),致病性信息源自国际通用基因变异解读分类标准
诊断:Evens综合征,原发性免疫缺陷病(STAT1基因变异),慢性皮肤黏膜念珠菌病,甲状腺功能减低症。
治疗过程:患儿因慢性皮肤黏膜念珠菌感染及甲状腺功能减低症,曾接受长时程抗真菌治疗及外源性甲状腺素补充,目前三碘甲腺原氨酸、甲状腺素水平正常,TSH升高,身高有明显增长,但仍处于同龄儿童正常身高值第三百分位以下。本次入院考虑既往丙种球蛋白,激素治疗虽有效,但不能维持。加用免疫抑制剂西罗莫司0.5 mg/d调节患儿Treg细胞比例,血药浓度控制在9~12 μg/L,期间患儿血小板逐步上升至正常,未复发溶血,抗人球蛋白试验(+),网织红细胞比例降至2.25%,考虑患儿合并严重感染,不能长期大量应用免疫抑制剂,应用2个月西罗莫司后停药,停药后复查Treg细胞比例升至0.083。
功能获得型STAT1杂合变异常表现有慢性皮肤黏膜念珠菌、细菌或病毒感染,继发自身免疫病,淋巴细胞减少,脑动脉瘤以及肿瘤。本例患儿变异基因的致病性已经有文献报道,与皮肤黏膜念珠菌病相关[1],该变异不属于多态性变化,在人群中发生的频率极低。
本例患儿出生后即出现反复真菌感染,表现为鹅口疮,鼻窦炎,支气管肺炎,皮肤硬结等。生长发育严重迟滞,甲状腺功能严重低下,外源补充甲状腺素,TSH仍升高。患儿自身免疫性血细胞减少表现突出,反复溶血性贫血伴血小板减少。多次血常规检查示淋巴细胞绝对值偏低,淋巴细胞亚群提示NK细胞、Th17比例低,考虑可能为原发性免疫缺陷病。根据国内外关于原发性免疫缺陷病早期表现、早期诊断的专家共识[1,2,3],约50%有明确基因变异的X连锁免疫缺陷病患儿无家族史,且多数患儿为其单基因疾病新变异的第一批表现者,故基因变异检测是获得明确诊断的最可信的方法。对于大多数表现为全血细胞减少的患儿,早期紧急应用的免疫抑制治疗会干扰进一步相关的免疫学分析[4],对疑似患儿应在入院后及早进行临床免疫学相关检测。对于淋巴细胞绝对值减低、淋巴细胞亚群比例尤其是辅助性T细胞比例异常、Ig减低、补体异常的患儿应及早结合其临床表现进行原发性免疫缺陷病分组(共8组)排查[3]。通过分组排查结果可进一步锁定目标疾病群进行基因检测。本例基因筛查和测序结果证实,患儿自发STAT1基因杂合变异c.1154C>T(p.Thr385Met),国内尚无该位点报道。
本例患儿同时出现STAT1基因和AIRE基因变异,实属罕见。鉴于STAT1基因变异所致慢性皮肤黏膜念珠菌感染与AIRE基因变异所致自身免疫性多内分泌腺病综合征Ⅰ型(APS-1)的自身免疫性及感染的临床表现有所重叠,而患儿母亲虽有AIRE基因杂合变异却并未致病,且该位点c.1095+5G>A不属于外显子变异,致病性尚未见文献报道,故该患儿暂不考虑诊断为APS-1。
ADAMTS13基因是家族性血栓性血小板减少性紫癜的致病基因,为常染色体隐性遗传方式。患儿父亲该位点为杂合子,未发病。患儿为ADAMTS13基因杂合变异(NM_139025):c.347G>A(p.Arg116Gln),但临床检测尿常规潜血阴性,外周血涂片未见破碎红细胞,血管性血友病因子裂解蛋白酶活性正常,ADAMTS 13抗体阴性,不支持血栓性血小板减少症的诊断,排除该病可能。最终考虑患儿为功能获得型STAT1基因变异所致慢性皮肤黏膜念珠菌感染继发血细胞减少。本病目前尚缺乏有效治疗方法。文献报道5例病情严重并反复出现真菌、病毒感染的患者接受了异基因造血干细胞移植,3例于数月后分别死于噬血细胞性淋巴组织细胞增生症、感染和间质性肺病[5]。虽然真菌对唑类耐药严重,但长时程抗真菌治疗仍是首选,且应注意药物相互作用,如本例患儿联用雷帕霉素与唑类药物后雷帕霉素血药浓度大幅提高。其余治疗方式例如:重组人粒细胞集落刺激因子,JAK1/2抑制剂鲁索利替尼,重组IL-17A或IL-17F或STAT1抑制剂均在国际上有所尝试并报道[6] ,疗效仍有待进一步观察、证实。

























