
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿女,2日龄,因进行性呼吸困难1 d急诊入住郑州市儿童医院。患儿为第一胎第一产,母亲孕36+6周因"胎儿宫内窘迫,羊水少"实施剖宫产。患儿出生体重2 050 g,生后精神反应差,吃奶少,24 h未排胎便,36 h出现轻-中度黄疸。生后第2天出现呼吸困难。父母为非近亲婚配,母孕期健康。入院体格检查:体重1 950 g,身长47 cm,心率175次/min,呼吸72次/min,皮下脂肪菲薄,皮肤黏膜轻-中度黄染,颜面及颈部散在数枚脓疱疹,呼吸节律不规则,三凹征(+)。双肺呼吸音粗,心音有力,律齐,心率175次/min,心前区可闻及柔和收缩期Ⅱ级杂音。肝、脾未触及,四肢肌张力增高,肌力正常。实验室检查:血白细胞计数10.43×109/L,中性粒细胞0.724(计数7.56×109/L),血红蛋白106 g/L,红细胞计数2.94×1012/L,网织红细胞0.118,提示溶血性贫血。外周血涂片分析:杆状核粒细胞0.16,分叶核0.27,提示核左移,可见中毒颗粒,成熟红细胞大小不一,分类计数100个白细胞,未见有核红细胞。尿酮体阴性。血气分析示代谢性酸中毒,pH 7.04,二氧化碳分压(PCO2) 17 mmHg(1 mmHg=0.133 kPa),氧分压(PO2)125 mmHg,HCO3-4.6 mmol/L,剩余碱(BE)-24.2 mmol/L,乳酸2.2 mmol/L,血糖5.6 mmol/L。血清总胆红素241.5 μmol/L,间接胆红素165.2 μmol/L,直接胆红素76.3 μmol/L,天冬氨酸转氨酶48 U/L,丙氨酸转氨酶55 U/L,谷酰胺转肽酶8 U/L。C反应蛋白12.3 mg/L;血清降钙素原(PCT)0.77 μg/L。血培养无细菌生长。脑脊液常规、生化正常,脑脊液涂片染色及培养均阴性。患儿血型O型,Rh阳性;Coombs试验阴性。新生儿溶血病检查均为阴性,叶酸、维生素B12水平正常。此后,多次复查血常规、C反应蛋白(CRP)、PCT和血气分析,均提示贫血、感染及代谢性酸中毒。胸X线片示两肺纹理增多、模糊。心脏超声示卵圆孔未闭。临床诊断"新生儿呼吸窘迫综合征、新生儿败血症、代谢性酸中毒、溶血性贫血、新生儿脓疱疹、卵圆孔未闭" 。
入院后患儿贫血进行性加重,血红蛋白74~83 g/L,红细胞计数(2.09~2.44)×1012/L,输注悬浮红细胞后贫血暂时有所改善,血红蛋白一度升至100 g/L。持续代谢性酸中毒难以纠正,血液pH值7.04~7.27, BE -16.1~-24.2 mmol/L,乳酸1.0~2.5 mmol/L,奶量稍增加即见代谢性酸中毒加重。生后第5天血氨基酸及肉碱谱分析提示多种氨基酸降低;尿有机酸分析显示5-氧合脯氨酸显著增高(513.420 mmol/mol肌酐),乳酸显著增高(72.650 mmol/mol肌酐),3-羟基丁酸显著增高(161.450 mmol/mol肌酐),修订诊断为:氧合脯氨酸尿症、代谢性酸中毒、溶血性贫血、多脏器损害。
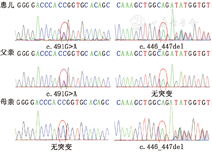
在知情同意的前提下,抽取患儿及其父母静脉血各2 ml,提取白细胞DNA,进行基因分析。参考文献[1]合成引物,对GSS基因的所有外显子进行PCR扩增,扩增片段包括内含子和外显子引物接头处,用LifeTouch基因扩增仪[TC-96/G/H(b)B]进行PCR-DNA扩增反应。PCR产物经纯化后测序,测序结果在NCBI-BLAST中进行序列对比(http://www.ncbi.nlm.nih.gov/BLAST/)。图1示患儿GSS基因第5外显子检出两处杂合突变,c.491G>A (p.R164Q)为父源突变,人类基因突变数据库报道为已知致病突变。c.446_447del (p.S149fs)为母源突变,未见文献报道,在人类单核苷酸多态性(SNP)数据库及千人基因组计划中均未见收录,200名正常对照中未检出。确诊患儿为谷胱甘肽合成酶(glutathione synthetas,GSS)缺乏症。
调整治疗方案:静脉点滴左卡尼汀、亚叶酸钙、碳酸氢钠、维生素C;肌肉注射维生素B12;口服维生素B1、E、AD;保证营养及热量供给、改善代谢。生后12 d死于多脏器功能衰竭。
GSS缺乏症(OMIM 266130)是一种罕见的常染色体隐性遗传病,以溶血性贫血、代谢性酸中毒、5-氧合脯氨酸尿症、神经系统损害为主要临床表现,严重者于新生儿期死亡[1],Li等[2]对5例中国患儿进行研究,发现差异表现显著[3]。本例患儿于生后2 d起病,严重代谢性酸中毒,伴进行性溶血性贫血、多脏器损害,经血氨基酸及尿有机酸分析发现氧合脯氨酸尿症,经GSS基因分析确诊。Hussain等[4]对41例患儿进行研究发现,病情的轻重与基因突变类型相关。本例患儿携带有移码突变及缺失突变,临床表型较重。本病需要终身治疗,大剂量维生素C、E可以减轻氧化应激损害,避免溶血危象[3]。苯巴比妥、磺胺类、阿司匹林等可诱发溶血危象,禁用于本病[3]。预后取决于病情轻重及早期正确诊断与治疗。对于新生儿期溶血性贫血和代谢性酸中毒的患儿,应考虑本病及甲基丙二酸尿症合并高同型半胱氨酸血症等有机酸代谢病[5]。先证者经基因确诊,下一胎可通过羊水5-氧合脯氨酸测定及基因突变分析进行产前诊断[5],对于降低发病率意义重大。

























