
1例主诉为"先天性巨结肠术后,智力发育迟缓"6月龄患儿就诊于首都儿科研究所附属儿童医院普外科。患儿3月龄时曾于本院行先天性巨结肠根治术,现因智力、运动发育落后复诊。经相关基因检测发现患儿携带ZEB2基因杂合无义变异c.756C>A(p.Y252X),国内外尚未见报道,且患儿父母均未携带该变异。结合患儿的特殊面容、临床表现、影像学检查及分子遗传学结果,诊断为Mowat-Wilson综合征。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿女,6月龄,因"先天性巨结肠术后,智力发育迟缓"于2016年9月就诊在首都儿科研究所附属儿童医院普外科门诊。患儿生后24 h内未自行排出胎便,需给予扩肛及开塞露帮助排出胎便。1月龄时因腹胀、呕吐在外院诊治,床旁超声显示动脉导管未闭、卵圆孔未闭、三尖瓣反流(具体不详)。3月龄曾以"出生后排便困难至今"为主诉收住我院普外科,下消化道造影显示患儿乙状结肠远端及直肠壶腹部呈痉挛状,乙状结肠近端以上结肠扩张,横结肠扩张,提示患儿先天性巨结肠(hirschsprung disease,HSCR),超声心动图显示患儿心脏无明显异常,接受本院普外科巨结肠根治术治疗,术中乙状结肠冰冻切片普通HE染色未找见神经节细胞。术后1周患儿无腹胀、呕吐,嘱依医嘱出院。患儿于6月龄时父母发现其智力、运动发育落后于同龄儿童,故来我院复诊。
患儿系母亲第1胎第1产,母亲产前检查无明显异常,患儿足月剖宫产出生,出生体重3.8 kg,身长51 cm,生后1 min、5 min、10 min的Apgar评分均为10分。父母均体健,否认近亲结婚,否认家族遗传病史。
入院体格检查:体温37.6 ℃,身高64 cm,体重7.5 kg,呼吸平稳,反应迟钝,腹部无明显腹胀,可见腹腔镜手术瘢痕;面容特殊,主要表现为眼距宽、眼眶深陷、低鼻梁、鹰钩鼻、小下颌(图1);无法抬头,无法独自翻身,不能独坐。
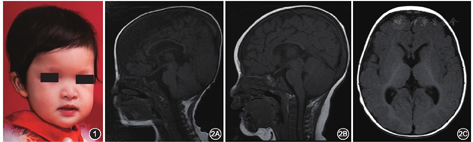
辅助检查:头颅磁共振成像(magnetic resonance imaging,MRI)结果显示胼胝体发育不良、白质髓鞘化延迟,侧脑室旁白质容积减少(图2)。
遗传学分析及诊断结果:结合患儿既往HSCR病史,特殊面容及智力运动发育落后,考虑患儿为综合征型巨结肠,为进一步确诊,建议患儿做HSCR相关基因检测。本研究经首都儿科研究所伦理委员会批准(伦理号:SHERLL2013039),患儿监护人签署知情同意书后,采取患儿及其父母外周血各5 ml,应用盐析法提取基因组DNA,送至北京迈基诺基因科技股份有限公司,针对172个HSCR及相关综合征致病基因的外显子及其侧翼序列(约100 bp)进行特异性捕获、测序。发现患儿携带ZEB2基因可疑无义变异c.756C>A(p.Y252X),Sanger测序验证及家系来源分析证实,患儿父母均未携带该变异(图3),提示为新生变异;查询人类基因变异数据库及PubMed数据库,该变异国内外均未见报道,为新变异。进一步查询ExAC数据库、gnomAD数据库和千人基因组数据库,未发现相同位点变异。ZEB2基因杂合无功能变异导致Mowat-Wilson综合征(Mowat-Wilson syndrome,MWS)的机制已经明确,该无义变异位于ZEB2基因N端,根据美国医学遗传学与基因组学学会指南本变异可判定为致病性变异,因此认为该患儿分子遗传学诊断明确,最终诊断为MWS。
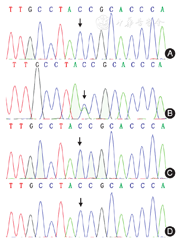
箭头所示为c.756C>A变异位置
治疗及随访:确诊后患儿一直在家由当地康复医院及患儿父母为其进行针对性的智力及行为发育训练,接受康复治疗,但疗效欠佳。至2018年4月患儿2岁2月龄,曾2次出现高热惊厥,有明显运动发育迟缓:嘴巴有不自主咀嚼动作,手臂不自主摇晃。能抓物翻身、靠背而坐、不能爬、不可直立。目前可听懂部分指令,表达好恶情感,无自主言语。
MWS是一种由ZEB2基因变异引起的罕见的常染色体显性遗传病,全球已报道了300多例[1],包含本例中国内地共报道2例[2]。国际上尚无具体诊断标准,导致临床诊断困难。本病临床表现多样,且与多种综合征存在交叉表型,MWS独特面容是识别本病最可靠的临床特征。300多例MWS患儿均表现面容特殊,最常见为小头畸形,其余有耳垂隆起、圆形鼻尖、鼻小柱突出、前额突出、眼眶深陷、眼距增宽、宽鼻梁、鹰钩鼻、小下颌;尽管大多数患儿的特殊面容在新生儿时期就已比较明显,但仍有部分患儿的面部特征随年龄增加而逐渐变化而显现出来[3];另外所有患儿均发现智力低下(轻重程度不一);常见的其他临床表现还有:癫痫发作、尿道下裂、先天性心脏病、身材矮小、先天性巨结肠、生殖器官畸形、便秘、先天性肾脏畸形等[1]。96%的MWS患儿可在MRI检查时发现异常,其中胼胝体发育不全最为常见,其他常见异常有海马异常,侧脑室扩大和白质异常(厚度减少、局部信号改变),另有脑室颞角增大、皮质畸形、后颅窝畸形、基底神经节变大及脑肿瘤等也可见于MWS患儿[4]。中国内地发现的2例MWS患儿均有明显的面容异常及合并HSCR。本例患儿MRI结果显示胼胝体发育不良、白质髓鞘化延迟、侧脑室旁白质容积减少。第1例中国内地患儿因严重的先天性心脏病,在做MRI检查之前死于严重心衰。智力低下、小头畸形及特殊面部特征等表现在MWS患儿尤其是新生儿期常不明显,但MRI多可见胼胝体发育障碍。MWS患儿面部特征随年龄增加逐步显现,但幼儿期依然可寻见端倪,需临床首诊医生在进行体格检查时细致观察。虽然MWS合并HSCR者在既往报道中只占44.2%[1],但中国内地的2例MWS患儿均合并有HSCR。因此,若患儿诊断为HSCR且面部异常同时合并智力发育异常时,临床医生应给予高度怀疑,可考虑行MRI检查。
1998年MWS的致病基因被定位于2q21-q23[5],后进一步被定位于ZEB2基因,又被称为SIP1或ZFHX1B[4]。ZEB2基因编码的蛋白是一种神经系统发育过程中重要的多功能调节因子。已报道的典型MWS患儿主要由ZEB2基因的大片段缺失、移码或无义变异导致。其中已报道的无义变异大多位于ZEB2基因的外显子8上。中国内地报道的2例MWS患儿发现的基因变异均为ZEB2基因无义变异(另一例为ZEB2 c.904 C>T,Arg302X)。若患儿临床表现不典型,ZEB2基因的筛查可作为诊断MWS的必要手段[6]。鉴于目前MWS仍无有效临床治疗手段,产前诊断成为阻断疾病发生的重要方法。Wilson等[7]于2003年回顾分析了MWS患儿的产前情况,发现2例患儿在孕期检查时均出现颈部半透明带增宽。另外由于胼胝体发育不全是唯一可以在产前检测到的MWS重要特征[4],因此在妊娠期常规检查时若发现胎儿存在胼胝体发育不良应予以高度关注。本例患儿父母的再生育风险评估可分为两种情况:如其中一方为生殖细胞嵌合体变异携带者,则再生育时后代发生该基因杂合变异的风险会显著提高,可通过产前诊断予以明确;如父母体细胞和生殖细胞均无变异,则再生育后代发生相同变异且患病的可能性极小(据既往报道推测,中国香港地区发病率为1/130 000,日本发病率为1/90 000[8]),但仍建议患儿父母于再次妊娠时重点关注胎儿的胼胝体和心脏发育情况,同时行ZEB2基因全序列检查以明确胎儿基因型。
本例MWS患儿以及其ZEB2基因的新变异c.756C>A(p.Y252X)丰富了MWS的变异谱。由于HSCR是MWS中的一个重要临床表现,且中国内地2例MWS患儿均以HSCR为首发症状收入普外科,希望通过对MWS的临床描述和总结能提高临床医生特别是小儿普通外科医生对本病的重视。MWS患儿在产前可发现颈部半透明带增宽、胼胝体发育不良等现象,为产前诊断提供证据。另外,先证者的分子遗传学诊断对于患儿所在家庭具有重要意义,是遗传咨询及患病风险评估的重要依据。
所有作者均声明不存在利益冲突

























