
1例10月龄男性主诉为"发热2周余,皮疹1 d"的患儿就诊血液科,患儿头发灰黄,局部皮肤色素脱失,肝脾肿大,EB病毒活动性感染,骨髓粒细胞见包涵体,全外显子测序发现,患儿存在LYST基因c.7645C>T(p.Q2549X)纯合变异,为未报道过的新变异,同时检出家系8位成员为携带者,呈常染色体隐性遗传,确诊为Chediak-Higashi综合征(CHS)。CHS属于溶酶体功能缺陷的原发性免疫缺陷病,预后不良,异基因造血干细胞移植可改善免疫功能缺陷。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿 男,10月龄,因"间断发热2周余,皮疹1d" 2016年11月就诊于安徽省儿童医院血液肿瘤科。患儿入院前2周余无明显诱因出现发热,呈中高热,热型不规则,伴少许咳嗽。就诊当地医院,予输液治疗3 d,未见好转,查肝功能提示肝酶高。后就诊本院感染科,考虑传染性单核细胞增多症,予抗感染,静注人免疫球蛋白及保肝降酶等治疗,体温正常,病情好转后出院。出院后1周患儿再次出现发热,入院前1天躯干部出现充血性皮疹,伴血象高,肝脾肿大,再次收住入院。患儿系其母第4胎第4产,足月剖宫产出生,出生体重3.3 kg。父母近亲结婚,大姐18月龄时因"发热、皮肤局部白化病、肝脾肿大、EB病毒活动性感染"等疑似噬血细胞综合征逝世;二姐、三姐表型正常。
入院体格检查:体温39.9℃,脉搏145次/min,呼吸35次/min,体重11.0 kg。神志清,精神尚可,躯干部见密集分布充血性皮疹,头发呈灰黄色,面颊部及双手背皮肤可见色素沉着,咽充血,扁桃体Ⅰ度,悬雍垂可见点状白色分泌物,颈部可触及肿大淋巴结,心肺听诊阴性,腹膨,肝脾肋下约4 cm,质中,神经系统检查阴性。
辅助检查:血常规,白细胞35.7×109/L,中性细胞0.117,淋巴细胞0.853,红细胞3.84×1012/L,血红蛋白97.0 g/L,血小板204.0×109/L。外周血异性淋巴细胞0.22。乳酸脱氢酶492.0 U/L。血清铁蛋白284.0 μg/L。肝功能,丙氨酸氨基转移酶758.0 U/L;天冬氨酸转氨酶333.0 U/L;甘油三酯1.61 mmol/L;纤维蛋白原1.63 g/L;EBV-DNA:4.17E+3 6拷贝/L;EB病毒抗体:衣壳抗体IgG(+)、衣壳抗体IgM(-)、核抗原抗体(+)、早期抗原抗体(+)。IgG 12.73 g/L;IgA 0.37 g/L;IgM 0.96 g/L;淋巴细胞亚群CD3+0.827,CD3+CD4+0.185,CD3+CD8+0.591,CD3-CD16+56+0.090,CD3-CD19+0.033。血尿遗传代谢疾病筛查正常。眼底检查示眼球震颤(-)、虹膜色素正常、视网膜色素减少。腹部B超示肝脾大。头颅核磁共振示脑实质内未见异常信号,大脑半球各叶沟回增深,双侧脑室稍扩张,胼胝体细。胸部CT示未见实质性病变。骨髓细胞学,骨髓增生活跃,粒系增生活跃,占有核细胞0.484,早期粒细胞胞质中可见大小不等的红色包涵体及空泡。红系增生活跃,占有核细胞0.234,淋巴细胞增生活跃,其中异性淋巴细胞占有核细胞0.022,全片见巨核细胞356个,血小板聚集分布。
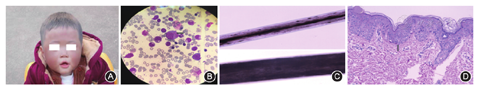
诊疗与随访:入院后根据患儿临床有发热,皮疹,咽峡炎,肝脾大,异淋升高,EB病毒阳性,初步考虑为传染性单核细胞增多症。但因患儿10月龄发病,局部有白化病,头发灰黄,局部皮肤色素脱失(图1A),视网膜色素减少,且骨髓粒细胞见包涵体(图1B)。光镜下患儿头发色素明显减少,呈小点状分布,非大块状分布(图1C)。电镜下患儿后背部皮肤活检示皮肤基底细胞水肿,真皮浅层血管周围有少量淋巴细胞浸润,黑色素颗粒异位缺失(图1D)。结合父母有近亲结婚,其大姐因类似临床症状逝世,临床考虑Chediak-Higashi综合征(Chediak-Higashi syndrome,CHS)可能。
征得患儿父母及亲属知情同意下,抽取患儿及其父母、姐姐、姑妈、姨妈、祖父母、外祖父母等家系成员共17人外周血各4 ml(EDTA抗凝),进行全外显子基因检测(由北京全谱医学检验实验室完成)。结果发现患儿LYST基因在第28外显子c.7645C>T(p.Q2549X)发生纯合变异,变异来自于父母。其他家系成员验证结果显示:患儿三姐、大姨妈、四姨妈、爷爷、外祖母和外祖母妹妹均为携带者,符合孟德尔常染色体隐性遗传。c.796G>T变异在dbSNP数据库、Hapmap、千人基因组数据库的分布频率为0,通过在线SIFT、Polyphen2软件预测蛋白结构结果有害。经检索文献Pubmed,ClinVar和万方数据库,该变异为未见报道的新变异。最终诊断为CHS。
该患儿入院后予阿昔洛韦抗病毒,甲泼尼龙琥珀酸钠抑制炎症反应及保肝降酶等治疗,病情好转。并于2017年2月行无关供者相合脐血干细胞移植,目前移植后2年余,血液和免疫系统的功能完全恢复。
CHS是一种罕见的常染色体隐性遗传病,属于溶酶体功能缺陷的原发性免疫缺陷病,发病率约1/100万。1943年由Beguez-Cesar首次报道,1952年Chediak报道了血液学特征,1954年Higashi描述细胞中存在特征性的过氧化物酶巨大颗粒,后被命名为CHS。
CHS是单基因遗传病,LYST基因是其唯一致病基因。1996年鉴定出LYST基因[1],定位于1号染色体q42.1-q42.2,基因组DNA全长205 878 bp,含53个外显子,mRNA全长13 493 bp;LYST基因编码的蛋白质为含3 801个氨基酸残基的胞浆蛋白(LYST蛋白)。LYST蛋白主要包含3个区域:含α螺旋的ARM/HEAT区、C端的BEACH区和WD-40重复序列区。
LYST蛋白功能目前尚未完全阐明,主要参与细胞内细胞器如黑素小体、溶酶体和其他细胞内分泌性颗粒的形成、结构和功能调节。CHS患者黑素细胞中黑素小体形成异常,引起色素减退,出现白化表现如皮肤色素减退,头发灰黄,畏光等;免疫细胞内溶酶体颗粒的异常,可导致NK细胞和细胞毒性T细胞活性缺乏,极易发生感染,尤其EB病毒感染或出现"加速期"表现,也即噬血细胞综合征;血小板中致密颗粒的异常,致血小板数量和功能异常而有出血倾向。神经元内溶酶体的异常,可引起神经系统的退行性病变如震颤麻痹、共济失调、癫痫、脑白质病变、痴呆等。按临床表现CHS分为三种表型:约80%~85%的患者为婴儿型,表现为婴幼儿期发生严重感染,出现加速期表现,大多于10岁前死亡。约10%~15%的患者为青少年型或成年型,其幼年期无明显异常,不会出现加速期,能长期存活到成年,但成年期可出现神经系统病变。本例患儿临床有反复发热,局部皮肤色素减退,头发灰黄,视网膜色素减少,肝脾肿大,EB病毒活动感染,骨髓粒细胞见包涵体,其大姐姐有类似症状而死亡,临床表型考虑为婴儿型。在临床工作中,如遇到反复感染的婴幼儿,同时具有白化病表现,一定要想到CHS可能,及时行血涂片找包涵体和基因检查有助于明确诊断。
从1996年LYST基因被首次报道以来,其变异热点区域尚不明确,基因型与临床表型的关系也尚不清楚。截止到2019年3月,国内报道了9例LYST基因变异的CHS患者,婴儿型占2/3(6例),共14个位点变异[2,3,4,5,6,7,8]。其中13个变异位点位于ARM/HEAT区,1个位于BEACH区。以移码变异(7个)和无义变异(6个)最常见。通常LYST基因移码变异、无义变异和剪切位点变异,更易导致蛋白质的结构和功能改变,临床表型更倾向于婴儿型。但也有报道LYST基因同一变异,也可表现不同的临床表型[9]。可见临床表型除了与基因型有关外,还有其他因素参与。本例患儿LYST基因在第28外显子发生c.7645C>T(p.Q2549X)纯合变异,其编码区7645位碱基C变异为T,导致编码蛋白质在第2 549位氨基酸(谷氨酰胺)处提前终止,由野生型的3 801个氨基酸变异为2 549个氨基酸的截短体蛋白。缺失了蛋白C端的BEACH区和WD-40重复序列区,其致病性可能由于触发NMD(无义变异介导mRNA降解),属于致病性变异。本例LYST变异与国内报道基本一致,变异位于ARM/HEAT区,为无义变异,临床表型为婴儿型。
CHS还没有根治的方法,80%患者于10岁内死亡,死亡原因为感染、出血和"加速期" (噬血细胞综合征)并发症。造血干细胞移植可改善血液和免疫系统的功能缺陷,使患者长期存活,但不能改善神经系统病变。造血干细胞移植5年无病生存率在60%~70%,影响移植的预后因素主要为供者类型和移植时机,同胞全相合供者或无关全相合供者,未发生加速期时移植效果好[10]。本例患儿在非加速期,接受了无关供者相合脐血干细胞移植,目前移植后2年余,血液和免疫系统的功能完全恢复,尚未发生神经系统病变。
报道1例CHS患儿的LYST基因在第28外显子c.7645C>T(p. Q2549X)发生了纯合变异,且为未报道过的新位点变异,临床表型为婴儿型。并检出患儿父亲、母亲、姐姐、姨妈、爷爷、外祖母等8人为该变异的携带者,符合常染色体隐性遗传。结果丰富了LYST基因变异谱,能为CHS的分子诊断提供依据。临床上如遇到反复感染的患儿,同时具有白化病表现,一定要想到CHS可能,行血涂片找包涵体和基因检查有助于早期诊断。
所有作者均声明不存在利益冲突

























