
1例主诉为"反复皮疹2个月、发热1个月余"的患儿就诊于深圳市儿童医院,曾被误诊为"结节性脂膜炎",经过基因及病理检查修正诊断为组织细胞吞噬性脂膜炎。本病是一种罕见的特殊类型脂膜炎,易继发噬血细胞综合征,临床上误诊率及病死率极高。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿 男,11岁4月龄,汉族。因"反复皮疹2个月,发热1个月余"于2018年6月入深圳市儿童医院。患儿入院前2个月无明显诱因出现双下肢暗红色瘀斑,家属未予重视。入院前1个月余出现反复发热,热峰39.5 ℃,热型不规则,双下肢瘀斑较前增多并伴压痛,当地医院诊断"结节性红斑",予阿莫西林舒巴坦钠治疗5 d,发热仍反复,骨髓涂片除外血液系统肿瘤后加用泼尼松40 mg/d口服,患儿发热缓解、皮疹渐消退,激素逐渐减量为30 mg/d,患儿再次出现发热,类型同前,下腹部及双下肢新发瘀斑,皮疹下可扪及痛性皮下结节,遂转至我院。患儿平素体健,个人史及家族史无特殊。
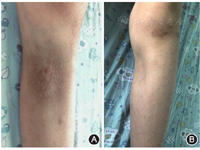
入院体格检查:体重30 kg,身高136.5 cm,体温36 ℃,心率115次/min,呼吸22次/min,血压111/65 mmHg(1 mmHg=0.133 kPa)。神清反应可,双下肢及左下腹可见散在浅褐色瘀斑(图1),直径2~8 cm,皮疹下可扪及皮下结节,直径2~4 cm,质硬,边界清晰,轻度压痛,局部皮温不高,浅表淋巴结无肿大,心肺查体无特殊,腹部稍膨隆,肝脏右肋下平脐,质中,边缘锐利,脾脏肋下及边,神经系统查体无异常,毛细血管充盈时间<2 s。
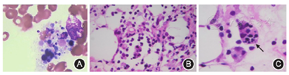
实验室检查:血常规示白细胞0.54×109/L,中性粒细胞0.09×109/L,淋巴细胞0.43×109/L,血红蛋白98 g/L,血小板44×109/L,网织红细胞26.40×109/L;尿常规、粪常规无异常;生化示丙氨酸转氨酶90 U/L,天冬氨酸转氨酶205 U/L,乳酸脱氢酶3 115 U/L,肌酐48.7 μmol/L,尿素氮3.9 mmol/L,肌酸激酶61 U/L;甘油三酯(空腹)3.67 mmol/L(参考值范围<1.7 mmol/L);铁蛋白29 900.4 μg/L(参考值范围22~322 μg/L);红细胞沉降率11 mm/1 h;凝血功能示纤维蛋白原0.57 g/L(参考值范围2~4 g/L);降钙素原0.13 μg/L,血培养、骨髓培养、EB病毒抗体IgM、EB病毒DNA、乙型肝炎病毒五项、丙型肝炎病毒抗体、人免疫缺陷病毒抗体、梅毒螺旋体特异抗体、结核菌素试验、结核免疫分析、真菌G试验、寄生虫检测及呼吸道病原检测等均未见明显异常。骨髓涂片提示骨髓增生极度减低,可见噬血细胞(图2A);肿瘤标志物阴性。Coomb试验阴性;抗核抗体、自身抗体谱、抗中性粒细胞胞浆抗体阴性;体液免疫示IgM 0.64 g/L、IgG 12.9 g/L、IgA 2.16 g/L;淋巴细胞亚群示总T淋巴细胞0.829、辅助T淋巴细胞0.507、抑制T淋巴细胞0.296、辅助与抑制T淋巴细胞比值1.72、B淋巴细胞0.153、NK细胞0.012、淋巴细胞计数467×106/L、总T淋巴细胞计数387×106/L、辅助T淋巴细胞计数235×106/L、抑制T淋巴细胞计数137×106/L、B淋巴细胞计数72×106/L、NK细胞计数6×106/L;细胞因子谱示白细胞介素(interleukin,IL)2 0.2 ng/L、IL-10 578.6 ng/L(参考值范围2.6~4.9 ng/L)、IL-4 0.1 ng/L、IL-6 9.9 ng/L、肿瘤坏死因子α 1.0 ng/L、γ-干扰素369.7 ng/L(参考值范围1.6~17.3 ng/L)。噬血相关检查示NK细胞活性14.19%(参考值范围≥15.11%);可溶性CD25水平1 127 ng/L(参考值范围<6 400 ng/L);信号淋巴细胞活化相关蛋白、X连锁凋亡抑制因子、穿孔素及颗粒酶B表达均无异常。皮下结节病理示脂肪小叶炎(图2B),可见"豆袋细胞"(图2C),未见肿瘤细胞;免疫组织化学示CD20(个别+),CD3(多数+),CD38(少量浆细胞+),CD4(部分+),CD8(部分+),TIA-1(部分+);EBER原位杂交阴性。
影像学检查:腹部超声提示肝脏弥漫性增大(右肋下平脐);心脏超声无异常;胸腹CT提示肝脾弥漫性肿大。全身正电子发射计算机断层显像检查无肿瘤表现。
基因检测:原发性噬血细胞综合征相关基因PRF1、UNC13D、STX11、STXBP2、RAB27A、CHSl/LYST、AP3β1、SH2D1A、BIRC4、MAGT1、Munc18-2、Rab27a等均未见突变;皮下结节基因重排检测提示TCRG(Vγlf、Vγ10-Jγ)阳性(+++)、TCRD(Vδ-Jδ、Dδ-Dδ、Dδ-Jδ)阳性(+);外周血T细胞、B细胞均未检测到单克隆重排。
诊断:根据患儿病理检查结果诊断为组织细胞吞噬性脂膜炎(cytophagic histiocytic panniculitis,CHP)。
诊治经过:入院后予甲泼尼龙800 mg/d静脉滴注治疗3 d,丙种球蛋白1 g/(kg·d)连续治疗2 d,头孢哌酮舒巴坦抗感染及对症支持治疗。采用噬血细胞综合征1994方案治疗,患儿症状缓解,噬血指标好转,激素规律减量,病情稳定后出院随访。
随访情况:患儿完成噬血细胞综合征1994方案的8周诱导治疗,定期监测噬血指标无异常,随访1年病情稳定。
CHP是一种罕见的脂膜炎,绝大多数患者为成年人,儿童CHP较为罕见,局灶型CHP多呈良性病程,而系统型CHP常伴多系统损伤并极易并发噬血细胞综合征,病死率极高。自1980年首次报道以来[1],目前已报道的儿童CHP仅23例,男女比例为1∶1.56,最小发病年龄为6月龄,中位起病年龄为16岁。所有患儿均有发热伴皮下结节,其他临床表现包括脾肿大(62%)、肝肿大(57%)及淋巴结肿大(33%),实验室检查特点包括肝功能损伤(89%)、凝血功能异常(82%)、贫血(81%)、血小板减少(76%)及白细胞减少(71%),几乎所有儿童CHP的骨髓象均可见噬血细胞。本例患儿以反复发热、肢端皮疹伴痛性皮下结节为主要表现,实验室检查提示血三系减低、铁蛋白升高、低纤维蛋白原血症及高甘油三酯血症,影像学检查提示肝脾弥漫性肿大,符合上述儿童系统型CHP的临床特点。本病的病理特征是形态学良性的T细胞和吞噬细胞浸润皮下脂肪组织,部分组织细胞吞噬完整血细胞或细胞碎片,形成特征性的"豆袋细胞"[2],本例患儿皮损组织病理呈典型CHP病理特点。
CHP的发病机制尚不明确,目前推测CHP可能是一种T淋巴细胞单克隆增殖性疾病,过度增殖的T淋巴细胞释放细胞因子风暴从而导致噬血细胞综合征发生。本例患儿细胞因子谱提示肿瘤坏死因子α、IL-10显著升高,提示其T淋巴细胞和巨噬细胞的过度活化[3]。本病的"良、恶"性质界定尚不明确,目前推测CHP可能为皮下脂膜炎样T细胞淋巴瘤(subcutaneous panniculitis-like T-cell lymphoma,SPTCL)自然病程的早期阶段[4]。本例患儿的病理特征及免疫组织化学虽与SPTCL不符,但患儿皮下结节基因重排提示TCR寡克隆基因重排,预示有进展为SPTCL的潜在风险。CHP的确诊主要依靠病理学检查。CHP在临床和病理上极易与结节性脂膜炎混淆,虽然两者的临床表现相似,但CHP多系统损伤突出,易出现血三系减低、肝功能受损或凝血功能异常;尽管两者的病理特征均可见皮下脂肪组织被组织细胞浸润,但CHP的组织细胞吞噬血细胞和细胞碎片,形成特征性"豆袋细胞",而结节性脂膜炎的组织细胞吞噬脂质,形成"泡沫细胞"。由于缺乏罕见病的病理诊断经验,本例患儿的组织病理被误诊为"结节性脂膜炎",经过临床医生反馈后最终修正诊断。
CHP尚无有效治疗方法,目前提倡糖皮质激素联合免疫抑制剂(如环孢素A、硫唑嘌呤、依托泊苷等)治疗[5,6]。目前已报道的23例儿童CHP中,病死率为17.39%,2例死于弥漫性血管内凝血,1例死于左心衰竭,1例死于消化道大出血。单用糖皮质激素治疗的缓解率为20%;糖皮质激素联合免疫抑制剂治疗的缓解率达88.24%,其中采用CHOP-E方案(环磷酰胺、长春新碱、阿霉素、泼尼松、依托泊苷)的缓解率为100%,糖皮质激素联合环孢素A治疗的缓解率为83%,最长随访35个月未复发;生物制剂及自体外周血干细胞移植成功治疗复发、难治性CHP亦有报道[7]。本例患儿在确诊CHP合并噬血细胞综合征后,采用噬血细胞综合征1994治疗方案,给予地塞米松联合依托泊苷诱导治疗8周,治疗效果理想,目前随访1年未复发,但应警惕其发展为SPTCL,临床上仍应严密随访。
所有作者均声明不存在利益冲突

























