
女,1岁3月龄,2月龄出现睡眠障碍,随后发现对疼痛不敏感、多汗、瘙痒、运动过多、自残、语言和运动发育迟缓等。体格检查营养不良体貌,可见多发软组织损伤。基因分析发现患儿SCN11A基因存在自发性杂合子变异(c.2432T>C, p.Leu811Pro),确诊为遗传性感觉和自主神经病Ⅶ型(HSAN Ⅶ),本病非常罕见。本例提示在婴儿早期出现睡眠障碍的时候,应注意HSAN Ⅶ可能,早期进行基因检测有助于诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
女,1岁3月龄,因“睡眠障碍13个月,搔抓11个月,运动过多伴自残7个月”于2019年5月在首都医科大学附属北京儿童医院神经内科住院治疗。患儿为其母第2胎第2产,足月自然出生,出生体重3.1 kg,头围34 cm。患儿2月龄时无明显诱因出现睡眠障碍,表现为入睡困难、睡眠片段化、易醒伴哭闹,每天睡眠时间尚可。患儿4月龄时发现出汗多,经常搔抓颈部,家长发现患儿对疼痛不敏感。患儿8月龄始频繁咬伤双手腕,并出现阵发性运动过多,表现为四肢舞动,身体扭动,烦躁、哭闹、大汗,持续数分钟自行缓解,每日多次,可连续15~20 d,间隔15~20 d再次出现,发作间期可完全正常。症状呈逐渐加重,睡眠时间明显减少,严重时3~4 h/d。1岁2月龄时患儿咬掉下唇及舌尖,齿缺失6颗。患儿自生后无明显喂养困难,排尿排便正常。患儿体重及身高增长慢,1岁3月龄时头围45 cm(第5百分位),体重8.2 kg(第3百分位以下),身高70 cm(第3百分位以下)。患儿语言和运动发育落后,9月龄会坐,1岁3月龄时不会爬、走,不会说话。曾就诊当地医院,诊断“癫痫、发育落后”,先后口服奥卡西平、氯硝西泮、托吡酯等疗效欠佳。患儿父母身体健康,非近亲婚配,家族中无类似病史。病例资料使用已获得患儿家长同意和北京儿童医院伦理委员会批准(编号:2020-Z-130)。
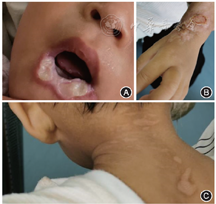
入院体格检查:营养不良体貌,脊柱正常生理弯曲,关节发育无异常,有眼神交流,无有意义语言,能听懂简单指令。能流泪,对温度变化敏感,不耐冷热。有触觉,无痛觉。图1可见多发软组织损伤(下唇部分缺如,舌尖缺如伴溃疡,颈后皮肤瘢痕,双手腕皮肤瘢痕及咬伤创面),牙齿缺如6颗,四肢肌张力偏低,肌力Ⅳ级,腱反射存在,病理征阴性,脑膜刺激征阴性。
辅助检查:入院后血红蛋白 77 g/L,存在小细胞低色素性贫血;血生化、甲状腺功能、乳酸、血氨、同型半胱氨酸、代谢病血尿筛查未见特异性改变;心电图和心脏彩超正常。胸部X线片正常。头颅CT及头颅磁共振成像双侧额颞部脑外间隙增宽,左侧脑室较对侧稍增宽。视频脑电图、肌电图、周围运动和感觉神经传导速度检查正常。
基因变异分析:获得父母知情同意后,采集患儿和表型正常的父母外周血样本,送检北京迈基诺医学检验所,通过全外显子测序及Sanger测序验证,发现患儿SCN11A基因第15外显子的新生杂合变异c.2432T>C(p.Leu811Pro),而父母均无此变异(图2),未发现其他与临床表现相关的基因。SCN11A基因的变异位点c.2432T>C(p.L811P)位于蛋白质的结构域Ⅱ中,并且通过多重序列比对分析在不同物种中高度保守。该变异在美国医学遗传与基因组学学会指南中提示有致病性。
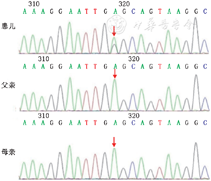
注:箭头示变异位点
诊断:根据患儿临床表现以及基因检测结果为SCN11A基因杂合子变异(c.2432T>C, p.Leu811Pro),确诊为遗传性感觉和自主神经病Ⅶ型(hereditary sensory and autonomic neuropathie Ⅶ,HSAN Ⅶ,在线人类孟德尔遗传号码为615548)。
治疗与随访:患儿住院期间给予氯硝西泮片 2 mg/d,硫必利片 25 mg/d,并给予补铁等对症治疗,症状有一过性缓解。出院后随访4个月,仍间歇有上述症状,改为维生素B1、B6各30 mg/d,甲钴胺0.5 mg/d,加巴喷丁100 mg/d口服,持续时间3个月,疗效欠佳停用。以后在症状加重时间断口服氯硝西泮1 mg/次,每天2次,对睡眠障碍有一定程度改善。患儿出院后已随访11个月,症状无明显改善,发作期基本呈持续状态,严重影响睡眠,白天及夜间均有烦躁、哭闹、肢体扭屈、抓挠;发作间期完全正常。患儿2岁1月龄会走,随访至2岁3月龄,仅会喊“妈妈”,有眼神及肢体交流,体重11 kg(第6百分位),身高84 cm(第3百分位以下),头围49 cm。
HSAN Ⅶ是由周围性感觉和自主神经元萎缩和变性引起的遗传性神经病[1, 2],由编码电压门控钠离子通道NaV1.9的 SCN11A基因变异所致,为常染色体显性遗传。截至2020年7月30日,已报道有3个SCN11A基因变异位点可导致HSAN Ⅶ型,分别是c.2432T>C(p.Leu811Pro)[1, 3, 4, 5],c.3904C>T(p.Leu1302Phe)[6, 7],和c.1187 T>C(p.L396P)[8],7篇英文文献报道了11个病例。SCN11A基因c.2432T>C(p.Leu811Pro)变异所致的HSAN Ⅶ型患者共报道了6例(包括本例患儿),男1例、女4例、性别不详1例,均具有明显的痛觉降低、多汗、生长迟缓;5例伤口愈合缓慢,瘙痒,胃肠疾病(肠胃功能紊乱、腹泻、便秘),对冷热不耐受;4例有无痛性骨折,肌张力低,轻度肌肉无力,颈后皮肤溃疡;2例排便或排尿时腹部、肛周或直肠阵发性剧烈疼痛,肌张力障碍姿势;其他临床表现还包括局部有痛觉、每日遗尿、伴有异位骨化的双侧髋部疾病、脊柱侧弯、哮喘、湿疹、胃食管反流、低血糖、维生素D缺乏症、头痛和手指皮肤刺痛[3];而认知功能、锥体系及锥体外系功能正常,本体感觉、振动觉等深感觉和温度觉则大致正常[1, 9]。与其他病例不同的是,本例患儿2月龄时就出现严重的睡眠障碍,主要表现为入睡困难、睡眠片段化以及睡眠哭闹,结合患儿随后出现搔抓及咬伤以及阵发性运动过多伴明显烦躁、多汗,推测以上症状与患儿存在严重瘙痒等感觉异常有关,需待患儿获得语言能力后进一步证实。Salvatierra等[3]也报道了1例SCN11A L811P基因变异的女性患者,虽然无皮肤病变如湿疹等,但其夜间瘙痒非常严重并且伴刺痛、出汗和下肢过度运动,导致入睡困难。由于已经报道的SCN11A L811P基因变异患者多数有严重瘙痒[4],推测与NaV1.9参与瘙痒机制形成有关。其次,已报道的HSAN Ⅶ型患者,虽然有运动和生长发育迟缓,但是智力正常。本例患儿存在明显的运动语言发育迟缓,是否与疾病本身导致的睡眠障碍、自主神经功能紊乱有关,尚不明确,需进一步随访观察。另外,本例患儿尚未出现骨折、骨骼发育异常等症状体征,而文献复习发现 HSAN Ⅶ型患者可在儿童早期反复出现无痛性骨折,骨折均愈合良好,骨骼的矿物质和生化指标正常,无放射学证据支持有骨病或骨质减少,骨活检显示骨组织结构正常[6]。由于轻微外伤就可导致骨折,仍提示骨质有非特征性缺陷,具体机制尚不明确[2]。尽管本病临床表现为周围性感觉神经病变,但由于HSAN Ⅶ型主要累及无髓神经纤维和小有髓周围神经纤维,大有髓周围神经纤维受累较少[6], 电生理检查往往无异常改变。本例及文献复习中患者的肌电图显示运动感觉周围神经的传导速度及波幅均正常,提示肌电图正常不能排除此类周围神经病[1, 3, 8]。本例患儿具有严重自毁容貌表现,痛觉明显缺失造成唇舌咬伤、皮肤溃疡瘢痕,伴有语言和运动发育迟缓,尤其需与Lesch-Nyhan综合征鉴别,该病是由次黄嘌呤-鸟嘌呤-磷酸核糖基转移酶缺乏引起的尿酸代谢异常的X连锁隐性遗传性疾病。Lesch-Nyhan综合征的特征是自残、舞蹈样运动障碍和智力异常[10],高尿酸血症是其临床特点,通过基因检测确诊。此外,HSAN还需要与各种获得性的周围性感觉神经病相鉴别。通常HSAN诊断取决于临床表现、特定的感觉和自主神经评估、病理检查以及基因变异鉴定[6]。因为大多数HSAN在临床和电生理诊断上非特异性[9],尤其是小婴儿期(<6月龄)起病,感觉障碍通常难于表述,容易造成诊断困难及延迟,因此进行基因诊断仍然至关重要。各型HSAN均以对症治疗为主[9,11],多学科联合管理非常重要[9,12]。无痛型HSAN尚缺少特殊治疗方法。
所有作者均声明不存在利益冲突

























