
患儿 女,8日龄,因“血小板减少5 d”于2020年4月收入北京儿童医院。血常规示血小板减少,外院予人免疫球蛋白治疗无效,血小板输注暂时有效,外周血及骨髓涂片可见巨大血小板,低浓度瑞斯托霉素诱导血小板聚集阳性,二代测序提示血管性血友病因子(vWF)基因c.3946G>A(p.V1316M)杂合突变,诊断为血管性血友病2B型。本例系自发性突变,我国尚无该突变位点报道。本病以预防出血为主要处理原则,患儿无活动性出血,病情平稳,未输注血制品(含成分血)。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿 女,8日龄,因“发现血小板减少5 d”于2020年4月收入北京儿童医院。入院前5 d发现患儿血小板减少,血小板计数波动于(56~22)×109/L,无出血表现,外院予新鲜冰冻血浆和人免疫球蛋白(剂量不详)输注后血小板仍进行性下降,予机采浓缩血小板1单位和免疫球蛋白输注后血小板升至88×109/L,入院前1天再次下降至32×109/L,血涂片可见巨大血小板,遂以“新生儿血小板减少原因待查”转入北京儿童医院新生儿科。患儿生后24 h曾出现颜面及躯干皮肤黄染,伴纳奶欠佳,呕吐、腹胀,血常规示血小板计数140×109/L,血生化示总胆红素升高,以间接胆红素为主,诊断为新生儿黄疸,予以蓝光照射等治疗后好转。患儿为其母第1胎第1产,38+2周胎龄自然出生,为小于胎龄儿,脐带部位无出血,出生后无缺氧、窒息、抢救史。家族史无异常。卡介苗和乙肝疫苗接种处无红肿和出血。
入院体格检查:体温36.5 ℃,心率150次/min,呼吸45次/min,血压75/40 mmHg(1 mmHg=0.133 kPa),体重2.88 kg,身长48 cm,头围34 cm。神志清,精神反应可,神经及大运动发育正常,颜面及躯干部皮肤黄染,采血处可见皮下瘀斑,余无出血表现,浅表淋巴结及肝、脾未触及肿大,心、肺、腹及神经系统查体无异常。
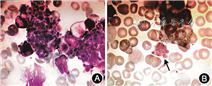
实验室检查(括号内为参考值):血常规示白细胞计数10.8×109/L(4.0×109~10.0×109/L),血红蛋白137 g/L(110~160 g/L),血小板计数12×109/L(100×109~300×109/L)。外周血涂片示可见大血小板,胞体大,可见聚集,血小板计数与仪器检测结果基本相符。凝血示凝血酶原时间12.4 s(9.4~12.5 s),部分凝血活酶时间24.7 s(25.1~38.4 s),纤维蛋白原0.94 g/L(Clauss法,2.0~4.0 g/L)。血生化示电解质、肝肾功能、乳酸脱氢酶等均在正常范围。住院期间血小板波动于(12~65)×109/L。粪常规示潜血阳性,红细胞3个/高倍镜视野。尿常规正常。骨髓细胞学检查示骨髓增生活跃,全片共找到42枚巨核细胞,偶见小巨核细胞,多数血小板体积偏大,可见巨大血小板和明显聚集现象(图1A)。小巨核细胞免疫酶标染色(CD41a)可见正常巨核细胞10个,大单圆核小巨核细胞3个,单圆核小巨核细胞4个,淋巴样小巨核细胞6个,全片巨核细胞23个(图1B)。抗血小板抗体、抗核抗体、抗双链DNA抗体、抗可溶性核抗原抗体谱阴性。血小板聚集率示二磷酸腺苷(adenosine diphosphate, ADP)、花生四烯酸和胶原刺激血小板聚集均降低,低浓度(0.5 g/L)的瑞斯托霉素诱导的血小板聚集(ristocetin-induced platelet aggregation, RIPA)阳性。血管性血友病因子(von Willebrand factor, vWF)抗原水平119.4%(66.1%~176.3%),凝血因子Ⅷ、Ⅸ、Ⅺ、Ⅻ促凝活性均正常。
基因检测:患儿vWF基因在外显子28存在c.3946G>A杂合错义突变(第1316号氨基酸由缬氨酸变为甲硫氨酸),经家系验证分析,患儿父母该位点均无突变,判定为自发性突变(图2),该vWF基因位点突变在人类基因突变数据库已有报道与血管性血友病2B型,即von Willebrand 病(von Willebrand disease, vWD)2B型相关,经生物信息学蛋白功能综合性预测软件REVEL预测结果为有害(SIFT、PolyPhen2、MutationTaster 和GERP+预测均为有害)。
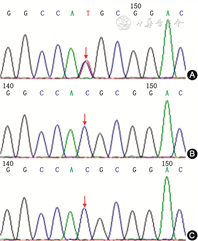
注:箭头所示为突变位点
根据患儿基因检测结果,结合患儿巨大血小板减少、血小板明显聚集和低浓度RIPA阳性,诊断为vWD 2B型。患儿确诊前血小板计数最低降至12×109/L,粪潜血阳性,予机采浓缩血小板0.5单位输注,2 h评估血小板增加数校正值(corrected count increment, CCI)=8.48,血小板回收率(percent platelet recovery, PPR)=18.3%,24 h评估CCI=3.52,PPR=7.6%,考虑输注有效,但不能维持,予以新鲜冰冻血浆50 ml输注,血小板维持在41×109/L,粪潜血转阴。出院后患儿无新鲜出血,血小板波动于(26~41)×109/L,未予特殊治疗,嘱防止磕碰和避免应用影响血小板功能的药物,观察随诊。
vWD是基因突变导致的vWF合成或功能障碍,是最常见的遗传性出血性疾病。其中vWD 2型主要为vWF功能的异常[1]。本例诊断为vWD 2B型,约占全部vWD发病率的5%,是由vWF基因A1结构域突变导致vWF与血小板膜糖蛋白Ib亲和力增加所致,这种“功能获得性”突变引起大分子量vWF多聚体降低甚至缺如,血小板过度聚集并消耗性减少[2]。已发现20种以上vWD 2B型的基因突变,本例患儿V1316M基因突变,也被称为“蒙特利尔血小板综合征”,是vWD 2B型的一种特殊类型[3],最易引起血小板减少,血涂片可见到血小板体积增大和异常活跃的血小板聚集,还会通过影响整合素αⅡbβ3信号通路导致血小板功能缺陷[4]。
本病呈常染色体显性遗传,多有阳性家族史,但目前已有关于V1316M自发突变的报道。vWD 2B型中30%~38%可发生不同程度的血小板减少[5]。有研究发现,V1316M基因突变与其他位点突变引起的vWD 2B型相比,更易出现严重的血小板减少,血小板体积更大,出血评分更高[3]。本例为V1316M自发突变,血小板呈中重度减少,血小板体积巨大,但患儿临床出血较轻,与文献报道不完全相符,仍需密切随访,警惕严重出血的发生。
本病临床上缺乏特异性,常被误诊为免疫性血小板减少症(immune thrombocytopenia,ITP),需要依靠特殊的实验室检查以协助诊断。如(1)低浓度RIPA阳性;(2)vWF瑞斯托霉素辅助因子活性(vWF∶RCo)与vWF抗原(vWF∶Ag)比值下降。本例患儿低浓度RIPA阳性,提示大分子量vWF与血小板表面膜糖蛋白Ibα亲和力增强,需考虑vWD2B和血小板型vWD(platelet-vWD, PT-vWD)两种疾病,后者主要是血小板GPIBA基因发生功能性突变,导致血小板表面异常的血小板膜糖蛋白Ibα与正常的vWF亲和力增加,产生与vWD 2B型相似的表型[6],改良RIPA或二代测序可用于鉴别vWF功能异常(vWD 2B型)和血小板受体异常(PT-vWD)[7]。本例患儿vWF: Ag正常,但由于本中心实验条件有限,无法进行改良RIPA和vWF∶RCo的检测,因此需要行二代测序协助诊断[1]。此外,本病还需与遗传性巨大血小板综合征(Bernard-Soulier syndrome, BSS)鉴别,但后者是主要发生在血小板GPIBA基因的功能缺失性突变,血小板膜糖蛋白Ibα无法与血浆中的vWF结合,因此在正常浓度和低浓度的RIPA阴性,可行鉴别。
本病治疗原则是预防出血,无活动性出血时尽量减少血制品(包括成分血)输注。出血时主要采用替代疗法,指南建议对于急诊手术和急性出血,应首选vWF∶RCo与vWF∶Ag比值接近1,经过纯化和病毒灭活的血浆来源vWF-FⅧ或高纯度vWF浓缩制剂[2]。止血方面可选用抗纤溶药物如氨基己酸和氨甲苯酸等。去氨加压素(1-desamino-8-arginine vasopressin, DDAVP)可通过刺激内皮细胞释放FⅧ和vWF达到止血作用,但对于vWD 2B型,DDAVP因会加剧血小板的消耗而被禁忌使用[2]。
综上所述,对于在生命早期出现血小板减少,特别是对免疫治疗效果不佳的患儿,应考虑到遗传性血小板减少症的可能,早期完善外周血和骨髓细胞形态学检查、vWF抗原及活性、血小板聚集率检测以辅助判断,如高度怀疑vWD 2B型,则需要进一步通过低浓度RIPA、改良RIPA和二代测序技术确诊并及时给予恰当治疗。
所有作者均声明不存在利益冲突

























