
患儿 男,1岁2月龄,因“阴茎短小、尿道下裂伴反复感染1年”就诊,合并发育迟缓、慢性腹泻、吞咽功能障碍、一过性血小板减少及贫血,结合基因检测,患儿7号染色体92732940位点存在c.2471G>A(p.Arg824Gln)的杂合变异,诊断为MIRAGE综合征。儿童MIRAGE综合征罕见。儿童期病死率高达62.5%,常见死因为呼吸衰竭、感染性休克、多脏器功能衰竭、失血性休克等。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿 男,1岁2月龄,因“阴茎短小、尿道下裂伴反复感染1年余”于2020年9月就诊于深圳市儿童医院内分泌科。患儿生后因“发现阴茎短小、尿道下裂、气促伴呻吟”于外院住院2个月余,诊断为“尿道下裂、新生儿肺透明膜病、支气管肺发育不良、早产儿、新生儿败血症”,予无创呼吸机辅助通气、肺表面活性物质替代,先后予多种抗菌药物抗感染治疗,外生殖器异常未予特殊处理。患儿3月龄后因反复呼吸道感染在本院就诊共13次,期间因血小板减少及贫血予对症治疗,定期随访血小板及血红蛋白维持正常水平。患儿添加配方后开始出现腹泻,6~7次/d,长期口服益生菌治疗。患儿3月龄后存在喂养困难,表现为反复呕吐及呛奶、并发“吸入性肺炎”,诊断为“吞咽障碍”,4月龄后长期鼻饲管喂养及进行吞咽康复训练。患儿1岁2月龄时可扶站、扶走,不能独走。仅发“baba、mama”音,能按指令完成动作。患儿因存在多系统异常,为明确诊断就诊深圳市儿童医院。患儿系其母第3胎第2产(因“葡萄胎”流产1次),出生胎龄31+5周,因“宫内缺氧”剖宫产出生,出生身长36.0 cm(-2.3 s),出生体重1 290 g(-1.3 s)。新生儿听力筛查通过。父母非近亲婚配,姐姐5岁体健,其余家族史无特殊。
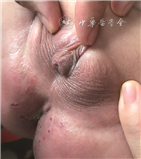
入院体格检查:心率115 次/min,呼吸30 次/min,血压89/46 mmHg(1 mmHg=0.133 kPa)。纠正胎龄后身长73.0 cm(-1.3 s),较出生时增加1.0 s,提示存在追赶性生长,体重9.2 kg(年龄别-0.8 s、身长别为均值),头围44.0 cm(-1.8 s)。神志清楚,精神反应可,前囟已闭。皮肤无色素沉着,无皮疹、出血点、瘀斑,弹性可。心肺腹及神经系统查体未见异常。四肢、脊柱无特殊。大耳朵,上腭高窄,双手通贯掌。外生殖器表现男性化不全,双侧睾丸容积1 ml、位于阴囊内,外生殖器男性化得分3分(图1)。
辅助检查:实验室检查示血、尿、粪常规未见异常;肝、肾功、电解质、心肌酶均正常。性激素(患儿3月龄时,化学发光法)示睾酮6.7 nmol/L,血清促卵泡刺激素1.830 U/L,血清促黄体生成素4.14 U/L,提示小青春期状态。抗缪勒管激素(化学发光法)及抑制素B(化学发光法)均正常。晨8点血清皮质醇(化学发光法)187.68 nmol/L。超声检查示男性生殖器超声示双侧睾丸位置、形态正常,大小均为1.3 cm×0.7 cm×0.6 cm(估算体积0.28 cm3);盆腔超声未显示子宫、卵巢声像;心脏、肝胆胰脾、泌尿系及肾上腺超声均未见异常。多次胸部影像学示肺炎。纤维支气管镜检示气管内膜炎症。心脏平扫及增强CT、头颅磁共振成像均未见异常。24 h咽喉pH监测示食管咽喉病理性酸反流。染色体核型为46,XY 22pstk+。
基因检测:经深圳市儿童医院伦理委员会批准(批准号:深儿医伦审[文章]2020(018)号)及患儿父母知情同意后,采集患儿及其父母外周血全外显子基因检测及家系验证。全外显子基因检测结果发现患儿SAMD9基因(NM_017654)第3外显子上存在1个杂合变异c.2471G>A,导致第824位氨基酸由精氨酸变为谷氨酰胺(p.Arg824Gln)(图2),人类基因变异数据库已有该位点MIRAGE综合征的相关性报道(与已鉴定的致病变异有相同氨基酸改变)。经家系验证分析,确认此变异为新发变异(经双亲验证的新发变异)。在正常人群数据库中未发现该变异,为低频变异(人群数据库中缺失)。根据美国医学遗传学与基因组学学会指南[1],该变异判定为致病性变异。SAMD9基因变异相关疾病为MIRAGE综合征,表现为骨髓发育异常、感染、生长迟缓、肾上腺发育不全、外生殖器畸形和胃肠道异常的一组综合征。
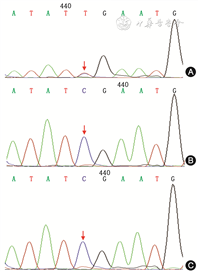
注:箭头所示变异位点
诊断及随访:患儿生后表现外生殖器异常,存在反复呼吸道感染、血小板减少及贫血、胎儿窘迫、出生小于胎龄儿、喂养困难、慢性腹泻,结合基因检查,该患儿7号染色体92732940位点存在c.2471G>A(p.Arg824Gln)的杂合变异,诊断MIRAGE综合征。患儿1岁5月龄时复诊,自出院后发生肺炎2次,多次痰培养为铜绿假单胞菌定植。校正胎龄后,患儿身长追赶至77.2 cm(-1.0s),年龄别体重追赶至10.5 kg(-0.4 s),头围44.0 cm(-2.3 s)。尝试自主进食后再次出现吸入性肺炎,予继续鼻饲喂养及吞咽康复训练,腹泻情况较前无变化。患儿可独走,会说“爸爸、妈妈、走”,会做“再见”。外周血血小板、粒细胞、淋巴细胞、血红蛋白均正常,外周血7号染色体荧光原位杂交,未检出7染色单体及7号染色体长臂缺失,未行骨髓细胞学检查。
MIRAGE综合征(OMIM:# 617053)于2016年首次由日本Narumi等[2]报道,描述以骨髓发育异常、感染、生长迟缓、肾上腺发育不全、外生殖器畸形和胃肠道异常为特征的一组综合征。该综合征是由SAMD9基因发生功能获得性变异所致,为常染色体显性遗传病。SAMD9是一种广泛表达的多域蛋白,为生长抑制因子,可抑制细胞的增殖和分化[3]。对文献中已报道的40例患儿进行临床表现分析[2, 3, 4, 5, 6, 7, 8],男女比例为3.44∶1。16例患儿具备全部六联征。39例为早产儿,平均出生胎龄为30周。35例患儿存在骨髓发育异常,表现为血小板减少、贫血、中性粒细胞减少、淋巴细胞减少和骨髓增生异常综合征。37例患儿存在反复感染,见于呼吸道感染、败血症、颅内感染、泌尿系感染等,病情反复且较严重。有出生资料的37例患儿均为小于胎龄儿,20例生后身材矮小,其中2例存在生长追赶。32例患儿诊断肾上腺皮质功能低下,其中20例患儿的尸检或腹部超声结果提示肾上腺发育不全。46,XY患儿存在不同程度的男性化不全,甚至为完全女性表型,因此临床上更容易被检出。胃肠道异常表现中31例患儿为慢性腹泻,另有结肠炎、胃食管反流病、食管狭窄和贲门失弛缓症。其他异常还包括胎儿窘迫、慢性肺疾病、全面发育迟缓、中枢神经系统异常、面部畸形(前额凸出、眼距宽、内眦赘皮、耳位低、小下颌)、骨骼畸形(指趾弯曲挛缩、脊柱侧凸、指趾甲发育不良)、先天性心脏病(动脉导管未闭、肺静脉异位引流、室间隔缺损)、泌尿系异常、自主神经功能紊乱(多汗症、体温调节障碍)、听力受损、甲状腺功能减退以及不同程度的语言、运动、智力发育迟缓。随访患儿生存情况,10例患儿因反复严重感染致呼吸衰竭、感染性休克、多脏器功能衰竭死亡,2例患儿因消化道失血致失血性休克死亡,12例患儿死因不详,1例死胎,死亡年龄在12(0~144)月龄,病死率高达62.5%。截至2020年10月15例患儿存活,存活年龄为15月龄~16岁,中位年龄为9岁。
MIRAGE综合征临床罕见,对于同时存在骨髓发育异常、感染、生长迟缓、肾上腺发育不全、外生殖器畸形和胃肠道异常的患儿,需要考虑此病。值得注意的是,宫内发育受限、生长迟缓、肾上腺发育不全、外生殖器畸形常作为其他综合征,如IMAGe综合征、Silver-Russell综合征等的表现之一,需综合临床特点及基因检查鉴别。MIRAGE综合征可累及多个系统,应进行全面评估。导致MIRAGE综合征的变异位点位于7号染色体长臂,而7号染色体长臂与骨髓增生异常综合征关系密切,对于确诊患者,需动态随访外周血细胞及7号染色体长臂变化情况。
所有作者均声明不存在利益冲突

























