
患儿 男,3岁1月龄,因“反复喉间痰鸣4个月”就诊于浙江大学医学院附属儿童医院呼吸科,经过胸部CT、腹部超声、骨髓常规检查,以及肺活检、基因检测,最终诊断为尼曼匹克病C2型、弥漫性间质性肺病。部分尼曼匹克病C2型患儿可能在婴幼儿期出现严重的肺部病变,但神经或精神系统无异常或仅有轻微症状,基因检测是重要的诊断工具。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿 男,3岁1月龄。因“反复喉间痰鸣4个月”于2017年4月就诊于浙江大学医学院附属儿童医院呼吸科。患儿4个月前出现喉间痰鸣,单声咳嗽,未至医院就诊,在家中间断口服头孢克肟(具体疗程及药物剂量不详),仍有喉间痰鸣。9 d前出现2次间断发热,体温波动在37.0~38.1 ℃,每次发热1~2 d,当地医院胸部X线片提示“支气管肺炎”,先后给予“阿奇霉素及头孢曲松”静脉滴注治疗1周,喉间痰鸣减轻,复查胸X线片示“肺炎征象无好转”,遂至浙江大学医学院附属儿童医院门诊就诊,拟诊“慢性肺炎”收住院。患儿2015年1月因“血小板减少”在浙江大学医学院附属儿童医院门诊就诊,骨髓常规示巨核细胞增多,产血小板功能差,涂片中找到大量泡沫状细胞,疑为尼曼-匹克细胞(图1A)。既往多次因“肺炎”住院,曾于2015年8月诊断“重症肺炎,呼吸衰竭Ⅱ型,间质性肺病”于浙江大学医学院附属儿童医院呼吸科住院治疗,查胸部CT示“两肺散在斑片影及细网格样改变”,此后多次随访,复查胸X线片未见好转。患儿系其母第3胎第3产,足月自然出生,出生体重2.8 kg,生后无窒息抢救史、无高胆红素血症病史。2岁内生长发育同正常同龄儿;3岁时发现走路不稳,说话不能成句。父母非近亲婚配。大姐21岁,体健;二姐患有“癫痫”,7岁时死亡(具体不详)。
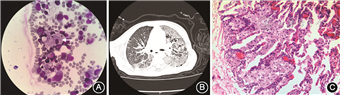
入院体格检查:体温37.3 ℃,心率122 次/min,呼吸48 次/min,血压99/60 mmHg(1 mmHg=0.133 kPa),神志清,精神一般,颈无抵抗,呼吸急促,轻度吸气性三凹征,两肺呼吸音粗,未闻及干湿啰音,心律齐,未闻及病理性杂音,腹软,肝肋下未触及,脾肋下1.5 cm,质地软。神经系统查体示双侧腱反射减弱,病理征阴性,四肢肌张力正常,肌力Ⅲ至Ⅳ级。
辅助检查:血常规示白细胞11.2×109/L,中性粒细胞0.60,嗜酸粒细胞0.02,血红蛋白133 g/L,血小板140×109/L,C反应蛋白2 mg/L。肝肾功能、过敏源及免疫球蛋白、T细胞亚群均未见异常。结核感染T细胞斑点检测、自身抗体20项、EB病毒抗体、巨细胞病毒抗体、1, 3-β-D葡聚糖试验、半乳甘露聚糖试验、呼吸道病毒免疫荧光7项、肺炎支原体RNA、痰细菌及念珠菌、曲霉菌培养、巨细胞病毒DNA均阴性。胸部CT示两肺大部呈磨玻璃状,内见支气管充气征及多发小囊状透亮区,小叶间隔增厚(图1B)。头颅磁共振成像示双侧脑室体后部斑片状长T2信号影,胼胝体偏薄,其余未见异常。腹部超声示脾脏肋间厚3.5 cm,肋下4.5 cm。入院第6天行纤维支气管镜检查及灌洗治疗,镜下见气管及各支气管管腔通畅,黏膜充血水肿,管腔内见少许白色黏稠分泌物,予生理盐水灌洗,洗出黏液栓样液体。入院第9天行胸腔镜下肺活检,病理示镜下见肺泡区域间隔增宽,泡沫细胞浸润及纤维组织增生,肺泡腔内见大片泡沫状细胞(图1C)。北京协和医院病理科会诊示(左上肺)肺泡腔及间质内见大量泡沫状细胞聚集,病变以小叶周边部位分布为主,部分间质增宽,病变符合脂质性肺炎,倾向内源性,需要除外先天性基因及代谢异常;免疫组化示AE1/AE3(-),CD163(+),CD68(+),S-100(+);糖原染色阴性。
诊疗及随访:住院后给予抗感染、吸氧、雾化吸入等治疗,住院30 d,患儿气促、咳痰减轻,胸部CT较前相仿,予出院。基因检测出院后结果回报14号染色体的NPC2基因外显子1存在纯合错义变异,表现为c.3G>A(编码区第3号核苷酸由鸟嘌呤变异为腺嘌呤),导致氨基酸改变 p.M1I(第1号氨基酸变异为异亮氨酸);经家系验证,患儿父母双方均检测到该位点相同的杂合变异(图2)。最终诊断为尼曼匹克病(Niemann-Pick disease,NPD)C2型;弥漫性间质性肺病。2017年12月患儿因“癫痫持续状态,呼吸衰竭,弥漫性间质性肺病,NPD C2型”再次入住浙江大学医学院附属儿童医院重症监护室,予呼吸机辅助呼吸。家属最终放弃治疗并拒绝随访。
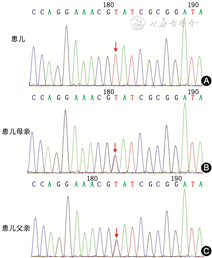
注:箭头所示为变异位点
讨论 NPD是一组罕见的致死性常染色体隐性遗传的神经内脏脂质沉积性疾病,分为A型、B型和C型。SMPD1基因变异导致酸性鞘磷脂酶缺陷,引起A、B型表现;NPC1及NPC2基因变异导致胆固醇转运及吞噬障碍,引起C型表现。NPDC型活产儿发病率为1∶150 000~1∶120 000,95%由位于染色体18q11.2的NPC1基因变异致病,仅约5%由染色体14q24.3的NPC2基因变异所致[1]。
NPD C2型根据累及脏器不同,临床表现多样。多累及神经系统,出现运动障碍、语言障碍、癫痫发作、精神问题等症状。肝脾受累表现为胎儿水肿、新生儿胆汁淤积、肝和(或)脾肿大等。而肺部受累通常局限于早发型或伴有严重NPC2基因变异的患儿,表现为婴儿期起病严重的呼吸衰竭,伴或不伴神经系统症状[1]。1994年,Steinberg等[2]报道了第1例死于呼吸衰竭的NPD C2型的患儿,表现为婴儿期不伴神经系统受累的早发型肝脏疾病及严重肺部疾病。此后,陆续有外文报道称少数NPC2基因变异的患儿可发生严重肺损伤、呼吸衰竭[1, 3, 4, 5, 6, 7, 8]。这些患儿肺、肝、脾病变大部分起病于婴儿早期(中位年龄3月龄),甚至新生儿期;起病后呼吸系统疾病迅速恶化,平均8月龄死亡;肺部影像符合弥漫性间质性肺病表现。近期,毛春婷等[9]报道了国内第1例有严重肺部疾病的早发型NPDC2型患儿,表现为婴儿期起病的反复呼吸道感染、呼吸窘迫,伴有胆汁淤积、肝脾肿大及精神运动发育的落后。本例患儿呼吸系统症状是整个临床过程中最显著的表现。1岁左右起病,主要表现为反复呼吸道感染和呼吸急促,胸部影像显示两肺弥漫性毛玻璃样改变。直至3岁,发现脾脏肿大、运动倒退及语言发育障碍,才被怀疑为NPD。
Ramirez等[10]研究表明,与正常小鼠相比,NPC2基因敲除小鼠肺内胆固醇合成及胆固醇含量均升高,且在其肺组织切片中发现大量含脂质的泡沫细胞。在本病例中,肺活检亦显示肺泡腔及间质内大量泡沫细胞聚集。此外,Griese等[5]和Yaman等[6]发现,部分NPD C2型患儿支气管肺泡灌洗液中肺泡表面活性物质显著增加。胆固醇代谢异常引起肺泡表面活性物质结构和功能异常,肺泡表面活性物质清除障碍,导致肺泡表面活性物质在肺泡积聚,最终导致肺泡蛋白沉积。本例患儿接受了纤维支气管镜检查及灌洗,但未发现肺泡蛋白沉积。
在已发现的78个NPC2基因变异中,有11个与肺部弥漫性病变有关。外显子1的 c.58G>T变异报道最多[1,3, 4],共有9例,其中8例c.58G>T纯合变异的患儿中,死亡7例,仅1例由于进行骨髓移植在5岁时仍存活。据此推测c.58G>T变异预后差。而3例c.82+2T>C纯合变异患儿中有2例存活[3,7, 8]。这些证据表明,NPD C2型患儿的呼吸系统表现和预后与基因变异类型可能存在相关性。本例患儿基因分析显示NPC2基因外显子1发生了c.3G>A纯合错义变异,在既往的文献中尚未见报道。
该病尚无有效治疗方案,对症治疗包括控制癫痫发作、改善肌张力障碍、胃造瘘等。美格鲁特是目前国际上唯一批准的用于改善儿童及成人NPD患者神经功能的药物,但是否能改善早发型NPD C2型患儿的肺部症状,尚待研究[11]。
总之,NPD C2型可能在新生儿期或婴儿期出现不伴神经系统症状的严重的弥漫性肺损伤。婴儿期弥漫性间质性肺病合并肝脏和(或)脾脏肿大,尤其伴有神经系统症状应提醒儿科医生意识到NPD C2型的可能性。基因检测是一种重要的诊断工具。不同基因变异类型的NPD C2型患儿的呼吸系统表现和预后可能各不相同。
所有作者均声明不存在利益冲突

























