
患儿 男,4岁主要临床表现为1岁后癫痫发作、发育迟滞和智力障碍。全外显子组测序提示两者在YWHAG基因的第2外显子均存在c.170G>A/p.R57H新的杂合错义变异,导致14-3-3γ蛋白第57位的精氨酸替换为组氨酸。患儿的c.170G>A变异遗传自母亲,而在外祖父母中未检测到,该变异为新自发变异,且在该家系内进行传递。患儿和母亲均为癫痫伴智力障碍,但临床高度怀疑发育性癫痫性脑病56型。


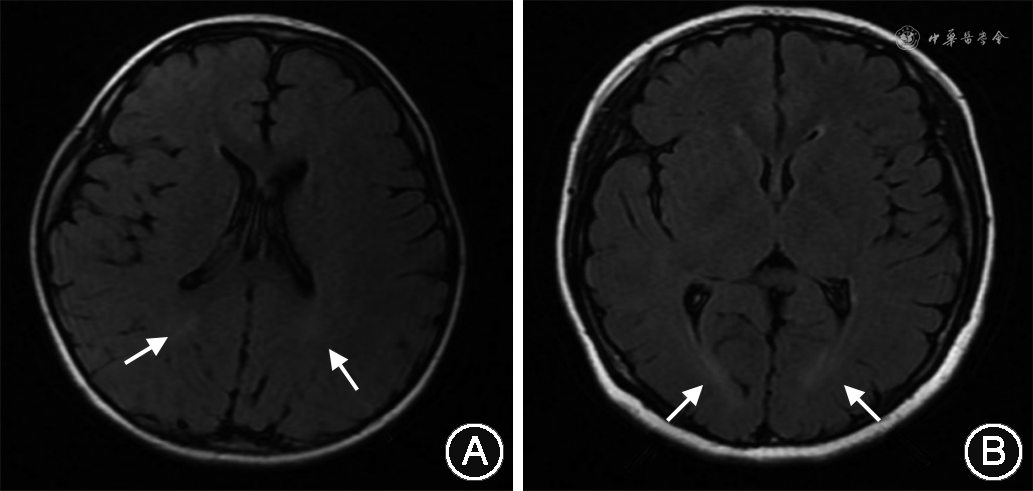
患儿 男,3岁3月龄,因“运动、语言发育落后1年余”于2020年3月至青岛大学附属医院儿童保健科门诊就诊,患儿就诊时仅会说“爸爸、妈妈、不要”等2字词语,表达意愿时仅会说“要”或手指示,情绪不稳定,容易发脾气,不会跳跃,自理能力欠佳。门诊行格塞尔发育量表示低于同龄正常幼儿,门诊考虑为“全面发育迟滞”。23月龄时无明显诱因出现抽搐1次,表现为双眼上视,口唇发绀,四肢抖动,呼之不应,无口吐白沫,无尿、粪失禁,持续1 min缓解。行颅脑磁共振成像(magnetic resonance imaging,MRI)未见异常,3 h视频脑电图未见异常,血常规、肝功能正常,血遗传代谢病氨基酸和酰基肉碱谱分析、尿液有机酸综合分析均未见明显异常。初步诊断为“癫痫”。给予丙戊酸钠3 次/d,4 mL/次,未再出现癫痫发作。患儿足月剖宫产出生,系其母第1胎第1产,出生时哭声响亮,Apgar评分不详,出生体重3 200 g。5月龄会抬头,6月龄会翻身,9月龄会坐,14月龄会爬,16月龄会走,2岁6月龄会喊爸妈。父亲(32岁)表型正常,母亲(30岁)有“癫痫”病史,1岁时出现痫性大发作,表现为双眼上翻,意识丧失,四肢抽搐,持续1~2 min缓解。服用丙戊酸钠250 mg,2 次/d控制效佳,服药5年后自行停药再次出现痫性发作,表现同前。口服“苯妥英钠50 mg,2次/d、卡马西平100 mg,2次/d”治疗,近2年无痫性发作。患儿母亲自幼智力障碍,未能上学。患儿母亲反应迟钝,记忆力差,Romberg征加强试验不稳,走直线不能。患儿母亲简易精神状态量表总分为9分,蒙特利尔认知评估量表总分为13分。心肌酶谱、肌电图、心脏超声均未见明显异常,颅脑MRI显示脑室周围白质异常信号(图1),脑电图未见异常。父母及外祖父母非近亲婚配。外祖父母和姨妈表型均正常。
4岁体格检查:体重16 kg(第25~50百分位数),头围50.8 cm(第50~85百分位数),身高98 cm(第3~10百分位数)。神志清楚,精神好,自主体位,呼名有应答,眼神交流欠佳。头颅无畸形,全身皮肤未见异常,伸舌无偏斜、震颤,颈部对称,软,眼球无震颤,双眼活动灵活,视力、听力正常。心肺腹查体、外生殖器外观和发育均无异常。颅神经无异常,四肢肌肉无萎缩、肥大,肌力及肌张力、腱反射均正常,双侧巴宾斯基征(-)、戈登征(-)、奥本海姆征(-)、霍夫曼征(-),颈软,克氏征(-),布鲁津斯基征(-)。
辅助检查:婴儿—初中学生社会生活能力量表为9分;不同年龄阶段格塞尔发育量表结果均显示异常(13月龄大运动发育商68,细动作发育商68,应物能发育商68,言语能发育商60,应人能发育商64;23月龄大运动发育商86,细动作发育商74,应物能发育商69,言语能发育商48,应人能发育商63;3岁3月龄大运动发育商53,细动作发育商48,应物能发育商51,言语能发育商46,应人能发育商51)。心肌酶谱、肌电图、心脏超声均未见明显异常,颅脑磁共振成像显示脑室周围白质异常信号(图1),脑电图未见异常。
分子遗传学结果:经患儿家属知情同意,青岛大学附属医院医学伦理委员会批准(伦理批件号:QYFYWZLL26230)。采集患儿及其他家系成员的外周静脉血,用血液基因组DNA提取试剂盒(北京天根生化科技有限公司)提取基因组DNA。Sanger测序验证表明,患儿及其母亲YWHAG基因(NM_012479.4)的第2外显子存在一处c.170G>A/p.R57H杂合错义变异(图2),导致蛋白第57位的精氨酸替换为组氨酸。根据美国医学遗传学和基因组学学会指南,该变异被初步判定为临床意义未明:PM2+PP3。该变异尚未见文献报道且人类基因突变数据库和ClinVar数据库均未收录该变异,表型正常的患儿的父亲(图2)、外祖父母和姨妈均未发现该变异,在100名无血缘关系的健康中国对照者中也未检测到该变异。患儿的c.170G>A/p.R57H杂合变异遗传自母亲,而在外祖父母中未检测到,因此,该变异为新自发变异,且在此家系内进行传递。结合患者的临床表型和分子诊断结果,患儿和母亲被诊断为癫痫伴智力障碍,但高度怀疑发育性癫痫性脑病(developmental and epileptic encephalopathy,DEE)56型。
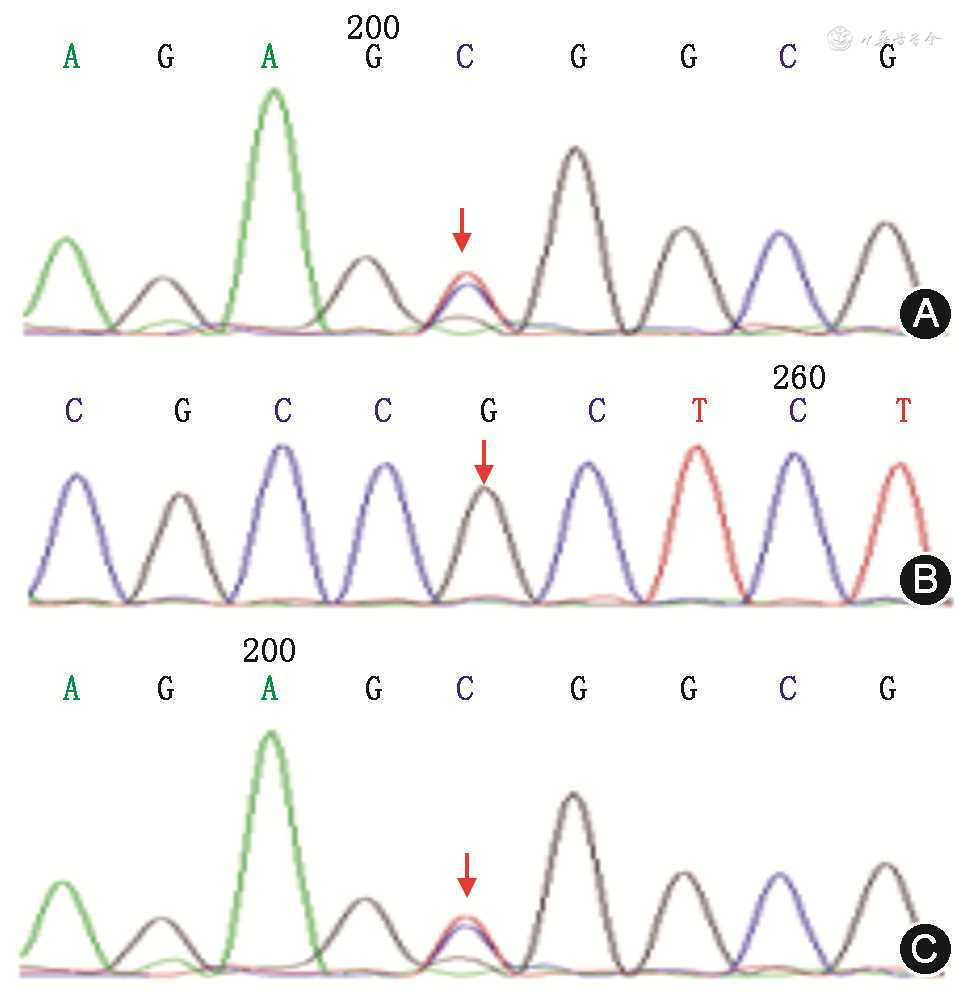
注:箭头示第170位的碱基
国际抗癫痫联盟分类和术语委员会于2010年对癫痫发作和分类框架术语及概念进行了修订,并对癫痫性脑病进行了定义,2017年将癫痫性脑病更新为DEE,又称早发幼儿癫痫性脑病,一类罕见的具有遗传异质性的神经系统疾病,以新生儿或婴儿期顽固性癫痫发作,脑电图异常,体格发育迟缓和智力障碍为典型特征。DEE伴爆发抑制由日本大田原及其团队于1976年首次报道并命名,描述了8例出生3月龄以内发病的患儿,故又名大田原综合征[1]。OMIM根据DEE致病基因的不同将其分为88种类型(DEE1~88型),其中以常染色体显性方式遗传的DEE56型(OMIM# 617665)是由YWHAG基因杂合变异引起的综合征。DEE的临床表型不一,从较容易控制的癫痫和轻度发育迟缓到药物抵抗性癫痫和严重的全身性发育迟滞。30%的DEE患者会在出生10 d内发作癫痫[2],且癫痫发作严重程度不一,发作过程可能为良性的,也可能为恶性的,严重者甚至会导致灾难性的大脑损伤[3]。Kanani等[4]在2019年描述了7例具有YWHAG基因杂合错义变异的患者,临床特征主要包括早发性癫痫,发育延迟和智力障碍并伴有孤独症。本例患儿及其母亲主要表现为1岁多癫痫发作、发育迟滞和智力障碍,此外,患儿也存在孤独症的风险,以上表型均符合DEE的临床特征。YWHAG基因与DEE56型的相关报道在世界范围内很少,Guella等[5]首次描述了4例DEE56型YWHAG基因自发变异的患者,临床表现为癫痫发作,发育迟滞和智力障碍,其中1例患者还伴有注意缺陷多动障碍,此表型与本例患儿相似。c.394C>T/p.Arg132Cys变异出现的次数最多,在3例患者中被检测到,只有1例患者具有c.44A>C/p.Glu15Ala变异,此外,Guella等[5]发现具有相同基因型的3例患者临床上癫痫的表型也较相似。Kanani等[4]在2019年在7例患者中发现了6个不同的自发杂合错义变异,其中c.394C>T/p.Arg132Cys变异在2例患者中被检出。c.394C>T变异可能为YWHAG基因的热点变异。在本研究中,该DEE患儿及其母亲YWHAG基因具有c.170G>A/p.R57H杂合错义变异,外祖父母未检出该变异,该位点也尚未见文献报道和数据库收录,表明这是一个新自发变异,但也不能完全排除罕见的外祖父母生殖细胞嵌合变异可能性。不同物种蛋白序列比对表明,14-3-3γ蛋白第57位的精氨酸位于蛋白高度保守区域。用HOPE软件(https://www3.cmbi.umcn.nl/hope/)预测变异对蛋白结构的影响,野生型14-3-3γ蛋白第57位氨基酸位于蛋白的核心区域,c.170G>A变异导致精氨酸替换为组氨酸进而可能干扰蛋白的核心结构。用PolyPhen-2、SIFT、Mutation Taster、PROVEAN和CADD软件对该变异进行生物信息学预测分析,得到的分值分别为0.999,0.000,1,-4.409和33,因此,该变异被评估为“有害变异”,具有较高的致病性。此外,家系验证表明YWHAG基因变异与癫痫伴智力障碍在该家系中共分离。结合临床表型和基因检测结果,YWHAG基因的c.170G>A是导致患儿及其母亲疾病的可能性致病变异,故被确诊为癫痫伴智力障碍,临床高度怀疑为DEE56型。在DEE致病变异的报道中,大部分患者都存在自发变异。如Singh等[6]在2015年描述了1例SCN8A基因c.3979A>G/p.Ile1327Val自发变异的DEE13型患者;Alagoz等[7]也报道了2例无血缘关系的土耳其男性DEE57型患者分别存在KCNT2基因 c.545A>T/p.Asn182Ile和c.2638C>A/p.Leu880Met新自发变异。DEE的诊断和分型主要根据基因检测和临床表型如早期难治性癫痫发作,异常脑电图,智力和运动发育迟缓。随着二代测序技术的发展,其高通量,快速,成本效益低等特点使其成为一种发现疾病候选致病基因和罕见病致病变异筛查的有效策略,随着越来越多的DEE被分型和命名,其基因型-表型的关系在未来工作中也将会被清晰地阐明。
所有作者均声明不存在利益冲突

























