
患儿 女,3岁,因发现身材矮小、指甲发育异常2年就诊。临床表现为身材矮小,指、趾末端发育异常,眼距增宽,鼻梁低平,空泡蝶鞍,小脑扁桃体下疝畸形,颅底凹陷,脊髓空洞,双肾囊肿。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿 女,3岁,因“发现身材矮小、指甲发育异常2年”于2021年4月就诊于西安大兴医院儿科。主要表现为身高增长缓慢,每年增长约3 cm,无关节肿痛,家属自行给予补锌治疗,效果欠佳。2年前家长发现患儿较同龄儿手指末端变软,指甲异常,无红肿、疼痛等不适。患儿平素23:00~24:00入睡,8:00起床,挑食,不喜蔬菜水果及肉类,运动适量,无应用补品及口服增高药史。前囟1岁半左右闭合,后囟1岁半到2岁左右闭合。1月龄时曾听力筛查未通过。患儿为其母第2胎第2产,足月自然出生,出生体重3.5 kg,Apgar评分不详,产后无窒息及抢救史,无产伤。生后足内翻。生后1周因“黄疸”在外院住院治疗(具体不详),母妊娠期甲状腺功能减低,口服“优甲乐”治疗,无感染发热史。生后混合喂养,3月龄可抬头,8月龄可稳坐,14月龄可稳走。体格发育较正常同龄儿稍缓慢。语言及智力发育同正常同龄儿童。父母体健,父亲身高172 cm,母亲身高162 cm,非近亲婚配,姐姐6岁3月龄,体健,否认家族中遗传代谢性疾病史。
入院查体:身高 91.5 cm(<P3),体重13 kg(<P3)。身材匀称,毛发粗糙浓密,颅骨无明显软化。头发浓密粗糙、前额突出,面部无黏液水肿,眼距增宽,鼻梁低平。心肺腹查体未见异常。脊柱、四肢外观无畸形,活动自如,左手食指、右手大拇指及食指指甲中央凹陷,边缘翘起,表面粗糙有条纹。余部分指甲表面粗糙有条纹。双下肢无畸形、水肿。肛门、外生殖器无畸形。四肢肌力、肌张力正常。皮肤感觉无异常,膝腱反射正常。
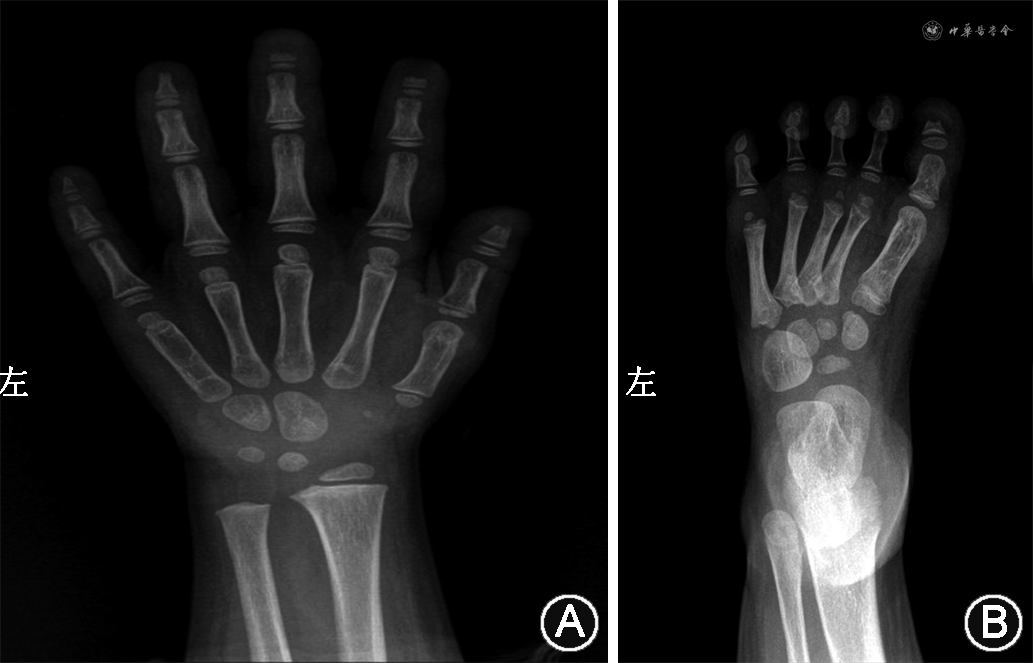
辅助检查:血常规、尿常规、肝肾功能、电解质、甲状腺功能、乙肝五项正常。生长激素激发试验示生长激素检测(空腹)1.3 μg/L;生长激素检测(30 min)4.0 μg/L;生长激素检测(60 min)9.3 μg/L;生长激素检测(90 min)9.9 μg/L;生长激素检测(120 min)3.7 μg/L,不完全缺乏。胰岛素样生长因子1 72.80 μg/L。微量元素六项示全血微量元素钙 1.60 mmol/L,镁 1.38 mmol/L,铁 7.53 mmol/L,铜 20.95 μmol/L,锌 68.86 μmol/L,铅 27.82 μg/L;甲状腺功能八项示三碘甲状腺原氨酸 2.51 nmol/L,余正常。空腹血糖 4.31 mmol/L;25-羟维生素D 67.0 nmol/L。骨密度示重度不足。心脏超声示心内结构及各心腔大小未见明显异常,左室收缩功能正常,彩色血流未见明显异常。泌尿系统超声示双肾囊肿。垂体磁共振成像示空泡蝶鞍,垂体高度约2.5 mm,垂体柄向右偏移;小脑扁桃体下疝畸形伴颅底凹陷,脊髓内异常信号影,考虑脊髓中央管扩张。胸椎、颈椎磁共振成像示Chiari畸形Ⅰ型(小脑扁桃体下疝畸形伴颅底凹陷,颈、胸段脊髓内异常信号影,考虑脊髓空洞);蝶鞍改变;胸椎多发蝴蝶椎。腕骨正位片示左腕骨5块,左腕骨龄符合3岁至3岁6月龄;左手第1~5指远节指骨部分骨质缺失,左足部X线片示第1趾末节趾骨部分骨质缺如(图1)。
基因检测:家系全外显子和线粒体基因检测提示NOTCH2基因 c.6709-6710insCACA(p.S2237Tfs*8)杂合移码变异,受检者父母和姐姐均未检出该变异,新生变异(图2)。
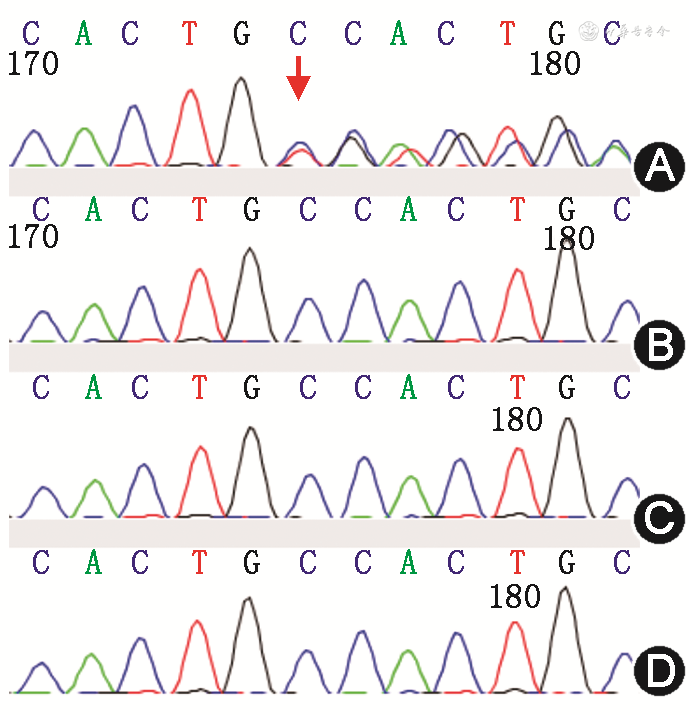
注:箭头所示为变异位点
Hajdu-Cheney综合征(Hajdu-Cheney syndrome,HCS)又称为遗传性骨发育不良并肢端溶骨症,主要特点是进行性的局灶性骨质破坏,包括肢端骨溶解和全身骨质疏松,可有身材矮小、面部异常、头颅肿瘤、听力丧失、先天性心脏病、肾囊肿等。最早分别于1948年和 1965年由3位放射科医生报道[1, 2]。该病罕见,全球报道约50 例,国内不足10例,为常染色体显性遗传,可有家族聚集性,也可为散发病例[3, 4, 5]。患者常常在青春后期或成人期因反复椎骨压缩性骨折而就诊,晚期生存质量和预后都很差。在儿童时期很少能看到HCS完整的临床表型,所以早期诊断非常困难,很容易误诊[4]。
HCS 的特征性表现为不同程度的骨丢失,包括指、趾末端肢端溶骨和全身骨质疏松。肢端溶解多从生后逐渐开始,在青春期和成人期进行性加重。据报道,该病对腰椎骨密度影响较大,而对股骨和桡骨的骨质影响较小。该综合征的临床表现为可变的表型[6]、广泛的临床表现和年龄依赖性进展[7, 8]。并且这些表现往往会随着时间的推移而演变,表型和症状逐渐恶化[7, 8]。HCS 最具代表性的临床表现为头颅畸形、颅缝闭合延迟、颅骨圆顶增厚、额窦缺失、蝶鞍拉长、下颌小、基底内陷、长头畸形和枕骨突出等颅骨改变;面部粗糙畸形、人中拉长、小颌骨、低位耳、浓眉、长睫毛、宽鼻子、高弓形的上腭、下颌错位、多毛症和眼距宽等面部改变;身材矮小、颈短、长骨骨折、关节松弛、双凹椎骨、脊柱后凸、颈椎不稳、椎体塌陷、膝外翻、蛇形腓骨、肢端骨质溶解、假杵状指、短指、进行性远端骨吸收、脱矿质、骨质减少和骨质疏松症等肌肉骨骼改变;先天性心脏病、动脉导管未闭等心血管改变;肠旋转不良等消化系统改变;脑积水和侧脑膜膨出等神经系统改变;尿道下裂、隐睾、肾囊肿和肾功能衰竭等肾脏改变;胸廓畸形、通气受限和反复感染等呼吸系统改变;运动发育延迟、听力丧失、声音变化、声音低沉、指甲短、足底溃疡和疝气等其他改变[6]。本例患儿也发现明显的骨质疏松,心脏结构无异常,有双肾囊肿。患儿身材矮小、眼距增宽、鼻梁低平、毛发粗糙、前额突出、下颌短小,指、趾末端肢端溶解。合并空泡蝶鞍,小脑扁桃体下疝畸形,颅底凹陷,脊髓空洞。据临床表现,实验室检查可初步做出HCS临床诊断。HCS 是一种由 NOTCH2 杂合变异引起的遗传疾病[9]。NOTCH 信号通路与机体骨骼发育和体内平衡密切相关 [10]。结合基因检测结果可早期明确诊断。本例患儿家系全外显子和线粒体基因检测:提示NOTCH2基因 c.6709-6710insCACA(p.S2237Tfs*8)杂合移码变异,患儿父母和姐姐均未检出该变异,为新生变异,该位点的变异在文献中尚未见报道。对HCS的病因研究非常有限,早期病例很多并无完整的基因检测结果,尚待收集更多病例的家系资料。尚无确定或有效的 HCS 药物治疗方法,尽管有报道双磷酸盐治疗可能有效[11],然而只有少数病例报告和评估了双磷酸盐治疗的有效性,其证据不足[12]。手术干预在某些情况下对并发症的治疗是有效的[13]。对 HCS 的治疗主要是对并发症和潜在风险的管理,以提高患者的生活质量和预期寿命。国外有报道给予患者阿仑膦酸钠治疗,但其有效性的证据不足。治疗的目的是缓解骨痛,减轻骨质疏松,提高骨密度,减缓肢端溶解的速度,从而降低胸、腰椎反复发生压缩性骨折的风险,提高晚期生存质量。但其治疗的远期效果,抑制肢端骨溶解的作用,仍有争议[11, 12],有待更多患者随访资料观察。建议本例患儿口服阿仑膦酸钠治疗,并加用维生素D,食物中适当补钙,每年监测骨密度和全身骨质溶解情况。
所有作者声明无利益冲突

























