
患儿 女,2岁6月龄,因“呼吸、心搏骤停后5 h”转入郑州大学附属儿童医院,主要临床表现为心搏骤停、晕厥、无自主呼吸、抽搐。心电图可见单发、成对室性早搏以及多形性室性心动过速,予持续机械通气、抗感染、脑保护、美托洛尔等治疗,遗留有言语和语言障碍、肌无力。基因检测结果提示患儿TRDN基因存在c.326delT(p.Leu109CysfsTer25)纯合变异,确诊为Triadin敲除综合征,后继续予美托洛尔治疗,随访7个月未发生心脏事件。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿 女,2岁6月龄,因“呼吸、心搏骤停后5 h”于2022年9月转入郑州大学附属儿童医院。患儿入院前5 h在睡眠期间出现呼吸困难、颜面及口周苍白,呼之不应,急诊至当地医院,发现患儿无自主呼吸及心跳,无发热、抽搐、呕吐等,立即给予心肺复苏术,后患儿心跳恢复,但无自主呼吸,给予气管插管及扩容、纠酸等治疗,治疗期间患儿出现抽搐。为求进一步治疗转入本院。患儿入院前1.5个月骑滑板车时摔倒,后枕部着地,出现神志不清、四肢强直、双眼上翻、尿失禁等症状,持续3~5 min自行缓解,当地医院行头颅CT示枕骨骨折,未予特殊处理。患儿为其母第1胎第1产,足月自然出生,围生期无特殊。生长发育同正常儿童。患儿父母非近亲婚配,家族中无特殊病史。
入院体格检查:体温37.5 ℃,脉搏133次/min,呼吸40次/min,血压105/64 mmHg(1 mmHg=0.133 kPa),血氧饱和度0.93。浅昏迷,对光反射迟钝,机械通气下口唇无发绀,颈软。心音低钝,律不齐,未闻及杂音。四肢肌张力正常,跟膝腱反射存在,布氏征阴性,双侧巴氏征阳性,双侧克氏征阴性。
辅助检查(括号内为参考值范围):血气分析示乳酸3.0 mmol/L(0.5~2.0 mmol/L),余正常;血常规示白细胞计数7.4×109/L(4.4×109~11.9×109/L),红细胞计数3.3×1012/L(3.5×1012~5.5×1012/L),血红蛋白92 g/L(112~149 g/L),血小板计数233×109/L(188×109~472×109/L),C反应蛋白21.93 mg/L(0~10 mg/L);肾功能正常;肝功能示丙氨酸转氨酶 87 U/L(8~30 U/L),天冬氨酸转氨酶 139 U/L(18~45 U/L);心肌酶示肌酸激酶同工酶21.7 μg/L(0~3.6 μg/L),B型脑钠肽前体1 222 ng/L(0~125 ng/L),肌钙蛋白T 0.142 μg/L(0~0.014 μg/L);电解质示钠 143 mmol/L(132~144 mmol/L),钾2.1 mmol/L(3.5~5.5 mmol/L),离子钙1.06 mmol/L(1.12~1.27 mmol/L),氯 103 mmol/L(98~108 mmol/L)。血氨基酸及酰基肉碱谱和尿有机酸分析未见异常。腹部B超未见异常。头颅CT示枕骨右侧线状透亮影。头颅磁共振成像示双侧大脑半球脑沟增深,双侧苍白球对称性异常信号。视频脑电图示弥漫性慢波活动。经颅多普勒示颅内大部分血管血流速度普遍减低,搏动指数及频谱形态未见明显异常。超声心动图示动脉导管未闭,左心室收缩功能稍减低。完善心电图提示窦性心律(V1~V4)T波倒置。完善动态心电图示室性早搏有3 151个,其中有2 343个单发室性早搏,50次成对室性早搏,52阵室性心动过速,45阵室性二联律,92阵室性三联律。
诊断及治疗:患儿入院后予以持续机械通气、抗感染、降颅压、脑保护、保肝、美托洛尔等治疗后心功能正常,复查心电图示早搏较前次减少,校正QT间期427~456 ms。药物治疗期间患儿未再出现心律失常,但肌力下降,神经反射差,遗留有言语和语言障碍,后转入我院康复科康复治疗。基因结果显示患儿TRDN基因存在c.326delT(p.Leu109CysfsTer25)纯合变异,患儿父母均携带c.326delT(p.Leu109CysfsTer25)杂合变异(图1)。根据2015年美国医学遗传学与基因组学学会发布的遗传变异分类标准与指南(American College of Medical Genetics and Genomics,ACMG),c.326delT(p.Leu109CysfsTer25)变异评级为致病性变异。根据基因结果以及临床表现,诊断Triadin敲除综合征(Triadin knockout syndrome,TKOS),继续予美托洛尔治疗,7个月后随访,未发生心脏事件,神经反射较前好转,肌张力仍低。
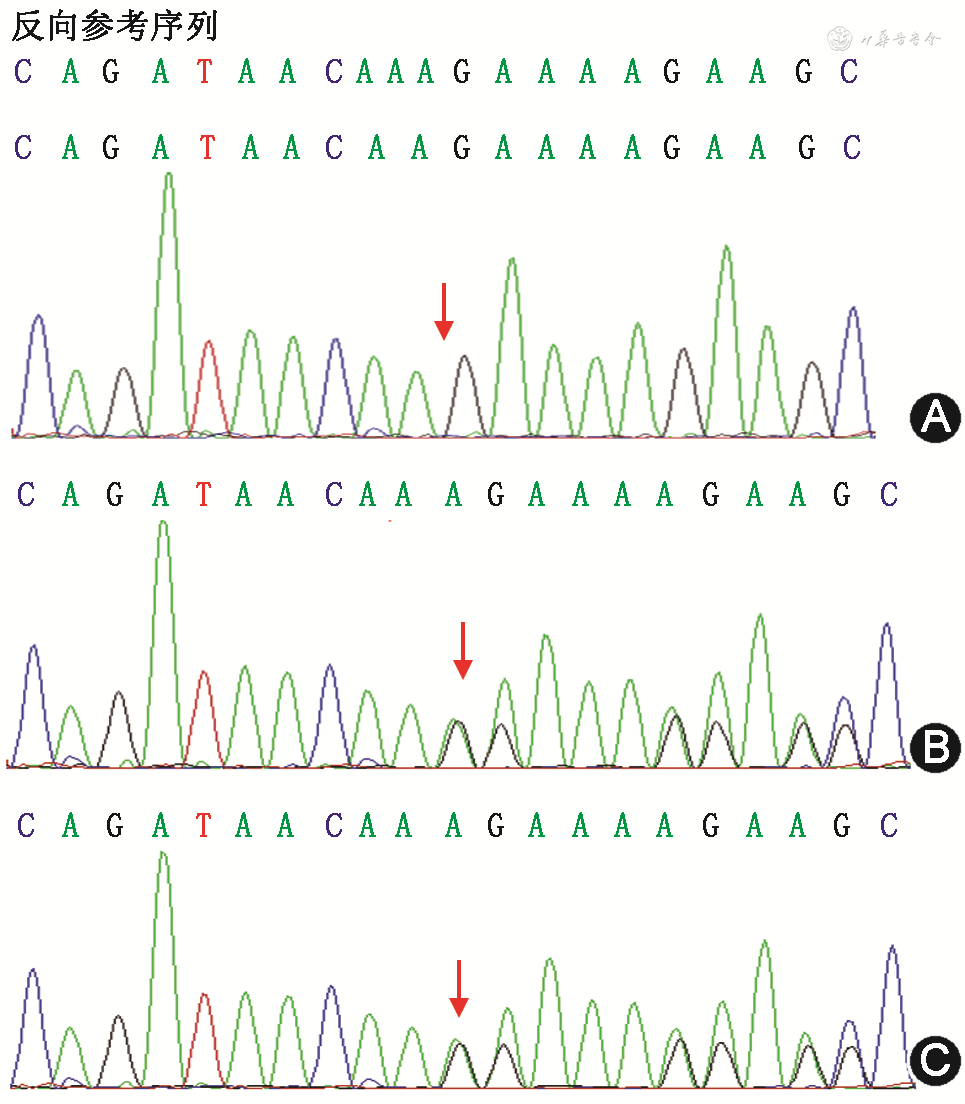
TRDN 基因定位于染色体6q22-q23,编码心脏钙释放单元复合体中的Triadin蛋白,该蛋白调控肌质网内Ca2+的释放,对心脏和骨骼肌正常生理功能起重要作用[1]。TKOS是一种罕见的遗传性心律失常综合征,由TRDN基因无效突变导致的Triadin蛋白功能丧失所致,遗传方式为常染色体隐性遗传[2],其临床表现为心搏骤停、晕厥、肌无力、心前导联T波倒置和短暂的QT间期延长等[2]。TRDN基因变异最早在儿茶酚胺敏感性多形性室性心动过速(catecholaminergic polymorphic ventricular tachycardia,CPVT)患者中发现[3]。2015年Altmann等[4]在最初被诊断为长QT综合征(long QT syndrome,LQTS)的5例患者中均检测到TRDN基因纯合或复合杂合变异,患者均表现出非典型性LQTS和CPVT特征,并且部分患者还表现出轻度至中度骨骼肌无力,Altmann等提出将TRDN基因变异导致的疾病命名为TKOS。
TKOS与LQTS、CPVT在表型上有一定的相似性,临床上易出现误诊的可能。TKOS患者发病年龄通常在5岁之前,比大多数LQTS或CPVT患者发病年龄较早[2]。TKOS患者在睡眠状态下可出现心搏骤停,静息心电图显示多态性室性心动过速、T波倒置和短暂性QT间期延长[2]。然而,典型的LQTS和CPVT分别以QT间期延长、运动或情绪诱发的多形性室性心动过速为特征。本例患儿临床表现为心搏骤停、晕厥、肌无力,心肺复苏术后静息心电图显示室早、多态性室性心动过速、V1~V4 T波倒置和短暂性QT间期延长,与既往文献报道的TKOS症状相符。基因检测结果提示患儿TRDN基因存在c.326delT(p.Leu109CysfsTer25)纯合变异,遗传自表型正常的父母,父母该位点均为杂合变异。根据ACMG指南,该变异可判定为致病性变异。结合患儿临床表现及基因检测结果,可诊断为TKOS。研究表明,Triadin功能丧失导致肌质网钙负荷增加和L型钙通道失活受损,这可能是导致TKOS患儿室性心律失常和肌力降低的原因[5]。
目前常规治疗手段如左心交感神经切除术以及β受体阻滞剂、氟卡尼和盐酸美西律等药物治疗不能有效地防治TKOS引起的恶性心脏事件,因此需要开发新的治疗方法用于治疗TKOS。蛋白质替代治疗可能是一种潜在的治疗手段。由于TKOS患者体内Triadin蛋白功能丧失,使用腺相关病毒血清型9将TRDN亚型递送到患者的心肌细胞,可能对治疗TKOS有效[2]。
综上所述,TKOS是以心搏骤停、晕厥以及肌无力为主要临床表现的高致死性心律失常综合征,对于临床上不明原因的心搏骤停、晕厥及心律失常的患儿应考虑到TKOS的可能,通过基因检测可确定诊断。
孙琪青, 王芳洁, 孙绘霞, 等. TRDN基因变异致Triadin敲除综合征1例[J]. 中华儿科杂志, 2023, 61(8): 735-737. DOI: 10.3760/cma.j.cn112140-20221221-01060.
所有作者声明无利益冲突

























