
先证者为5岁男童,自幼双眼内斜视,眼部检查示:双眼视杯宽而深,有多发睫状视网膜血管自盘周呈放射状发出,左眼可见视网膜神经上皮脱离。对先证者父母及妹妹行眼科检查,其父自述自幼右眼视力极差,10年前有肾移植病史。眼部检查示先证者父亲右眼视盘增大,血管走行异常,左眼视盘大小正常,多发视网膜血管自盘周放射状发出,可见黄斑区及视盘周围视网膜劈裂,右眼黄斑区视网膜神经上皮浅脱离。先证者妹妹双眼视杯宽而深,血管走行大致正常,黄斑区无明显异常。肾脏彩超及尿常规检查示先证者及其妹妹均可见双侧肾脏钙化灶及肾盂分离,轻度蛋白尿。基因检测结果示:先证者及其父亲、妹妹均存在PAX2基因c.419_421delGGA杂合突变。结合上述检查结果,诊断为:肾-视神经乳头缺损综合征。(中华眼科杂志,2021,57:454-457)


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先证者男性,5岁,因自幼双眼内斜视于2019年2月27日就诊于吉林大学第二医院眼科。眼部检查:右眼0.25矫正不提高,左眼0.4矫正至0.5。双眼前节未见明显异常。散瞳检查可见左眼视盘大于右眼,视杯宽而深,有多发睫状视网膜血管自盘周呈放射状发出,类似牵牛花视盘(图1)。左眼可见视盘颞上、颞下方圆形视网膜脱离。相干光层析成像术(optical coherence tomography,OCT)检查结果可见视盘宽而深,左眼可见视网膜神经上皮脱离,双眼黄斑区未见明显异常(图2)。询问其家族史,其父自述自幼右眼视力极差,10年前行肾移植手术,遂对先证者父母及妹妹行眼科检查。
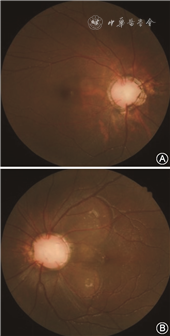
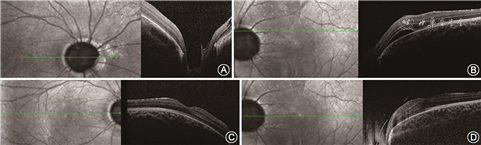
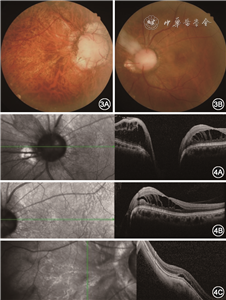
其母未见明显眼部异常,父亲视力为右眼20 cm手动,矫正不提高,左眼0.4矫正至0.8,前节无明显异常,散瞳查眼底可见右眼视盘增大,血管走行异常,盘周可见环形萎缩弧;左眼视盘大小正常,视网膜血管数目增多,自盘周放射状发出,可见视盘鼻侧有髓神经纤维及黄斑区视网膜劈裂(图3)。OCT同样可见视杯深而宽,左眼黄斑区及盘周视网膜劈裂,右眼黄斑区视网膜神经上皮浅脱离(图4)。先证者妹妹双眼矫正视力为0.2,眼前节未见明显异常,散瞳查眼底可见双眼视盘大小、血管数量及走行大致正常,视杯宽而深,颞侧盘沿窄(图5)。OCT可见宽而深的视杯,黄斑区未见明显异常(图6)。
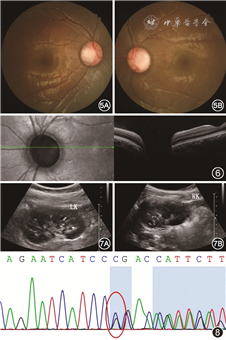
肾脏B超示先证者及其妹妹双侧肾脏钙化灶及肾盂分离(图7),患者父亲移植肾未见明显异常。先证者及其妹妹尿常规示轻度蛋白尿。采用目标区域捕获+高通量测序对先证者及其父亲、母亲、妹妹进行基因检测,在先证者及其父亲、妹妹PAX2基因外显子区域发现一处杂合突变:c.419_421delGGA(缺失突变),突变导致氨基酸改变p.R140_T141delinsP(缺失-插入突变)(图8),PAX2基因与局灶性节段性肾小球硬化症7型、肾-视神经乳头缺损综合征相关,为常染色体显性遗传。结合基因检测结果以及特征性的眼部、肾脏改变,可确诊患儿及其父亲、妹妹为肾-视神经乳头缺损综合征。
肾-视神经乳头缺损综合征是一种具有特征性眼部病变和泌尿系统病变的疾病。眼部病变主要包括视盘或视神经的发育不良、脉络膜、视网膜及虹膜的缺损以及视网膜脱离,部分患者存在近视或斜视。泌尿系统病变常表现为膀胱输尿管反流以及局灶节段性肾小球硬化,部分患者可出现蛋白尿[1]。除眼部及泌尿系统病变的症状外,高频听力丧失以及皮肤关节的松弛也有报道[2],在本家系中未发现听力及皮肤关节异常。
本家系发现位于常染色体上的PAX2基因发生c.419_421delGGA基因杂合突变(缺失突变)导致氨基酸改变 p.R140_T141delinsP(缺失-插入突变),此突变位点在HGMD pro数据库中未见报道。PAX2基因位于10q24.31,由13个外显子组成,编码蛋白转录因子,PAX2基因突变可导致视神经缺损和肾脏发育不全。在以往发现和报道的确诊肾-视神经乳头缺损综合征患者的中,仅有一半的患者可检测到存在PAX2基因突变[3]。已确诊的PAX2突变病例中约有92%的患者存在肾脏病变,77%出现眼部异常。只存在眼部病变或者肾脏病变的病例也有报道。PAX2基因突变的患者与无PAX2基因突变的患者相比,常表现为更严重的肾功能损伤以及视盘,视神经的缺损[4]。
肾-视神经乳头缺损综合征的表现极为多变,基因型与表现型之间难以建立明确的联系,即使是同卵双胞胎患者,眼部改变及肾功能改变仍存在巨大差异[5]。在视神经改变方面,有报道的患者群体中从轻微的视盘发育不良到牵牛花状视盘均有存在[6]。而在肾功能方面,部分患者很快发展为终末期肾病,需要透析甚至肾移植治疗,而有的患者仅表现为轻度或者一过性的蛋白尿[1]。
本次报道的家系中基因型相同,但表现型差异较大,这与既往报道中的基因型与表现型间关系不明确相符[5]。先证者及其父亲和妹妹表现为不同程度的视神经缺损,先证者父亲双眼视神经缺损程度及血管走行也不相同,视盘缺损较重,视盘增大且血管形态迂曲的右眼只有手动视力,而视盘缺损较轻的左眼可矫正至0.8。先证者及其妹妹在现阶段只表现为肾脏的钙化灶及轻度的蛋白尿,虽然其父在成年前已进展为终末期肾病,但是根据本病特征及以往的病例报道情况来看,并不能预测先证者及其妹妹未来是否会进展为终末期肾病。本病针对眼部改变长期随访的病例报告较少,在以往报道的随访时长约40年的病例中,患者唯一有视觉功能的眼睛没有表现出明显的视力下降[7]。视盘缺损会造成视网膜脱离风险增加,是患肾-视神经乳头缺损综合征患者除先天视盘及视网膜缺损以外视力严重受损的重要原因[6],需密切随访,早期发现并及时处理视网膜脱离,避免视力进一步受损。
对于基因检测结果为阳性的患者,长期的随访观察及生活方式的调整是避免肾功能进一步恶化以及视网膜脱离影响视力的重要手段。虽然在患有此病的家系中行产前基因检测可以确定胎儿是否带有突变基因,但是由于无法确定表现型与基因型的关系,难以判断携带突变基因的胎儿未来是否会面临严重的低视力以及肾功能损伤,所以产前基因检测在优生优育方面的作用极为有限。
首都医科大学附属北京同仁医院北京同仁眼科中心彭晓燕教授对本文的帮助
所有作者均声明不存在利益冲突

























