
患儿女性,2岁7个月,因左眼闭合不全、畏光1个月就诊于西京医院眼科。患儿行走不稳,面部不对称,双侧耳廓畸形伴耳聋;左眼内斜视、双眼屈光参差;左侧面神经麻痹;心脏彩色超声示动脉导管结扎术后。基因检测结果显示CHD7基因c.3392T>C。诊断为CHARGE综合征。(中华眼科杂志,2021,57:618-620)


患儿女性,2岁7个月,因左眼闭合不全、畏光1个月,就诊于空军军医大学西京医院眼科。患儿出生后哭闹时嘴向右侧歪斜,面部及耳部不对称,未予重视;2月龄时症状加重,于北京某医院诊断为“面神经麻痹”,未行特殊治疗;后家长发现患儿发育迟缓,6月龄时仍不会翻身、抬头;7月龄时患儿因“感冒后呼吸困难”,在当地儿童医院诊断为:(1)支气管肺炎;(2)心功能不全;(3)先天性心脏病(房间隔缺损中央型、动脉导管未闭、肺动脉高压);(4)精神运动发育迟缓;(5)左侧面神经麻痹;(6)左侧耳廓畸形。经对症治疗病情稳定后,至阿拉伯联合酋长国某医院行心脏手术治疗,具体术式不详。1个月前因患儿言语迟滞、行走不稳,于广东省某儿童医院诊断为:(1)运动障碍(运动发育迟缓);(2)精神发育迟缓;(3)先天性耳廓畸形;(4)面神经麻痹;(5)感音神经性耳聋(左耳极重度)。建议其验配助听器并行康复治疗,未执行。近1个月家长发现患儿睡眠时左眼闭合不全,伴畏光,为诊治眼部病变就诊于我科。患儿系第2胎第2产,足月顺产,出生体重2 900 g,新生儿评分不详,否认围产期特殊病变及生后吸氧史,否认先天性及家族性遗传病史。患儿父母健康,否认近亲结婚,患儿姐姐身体健康。
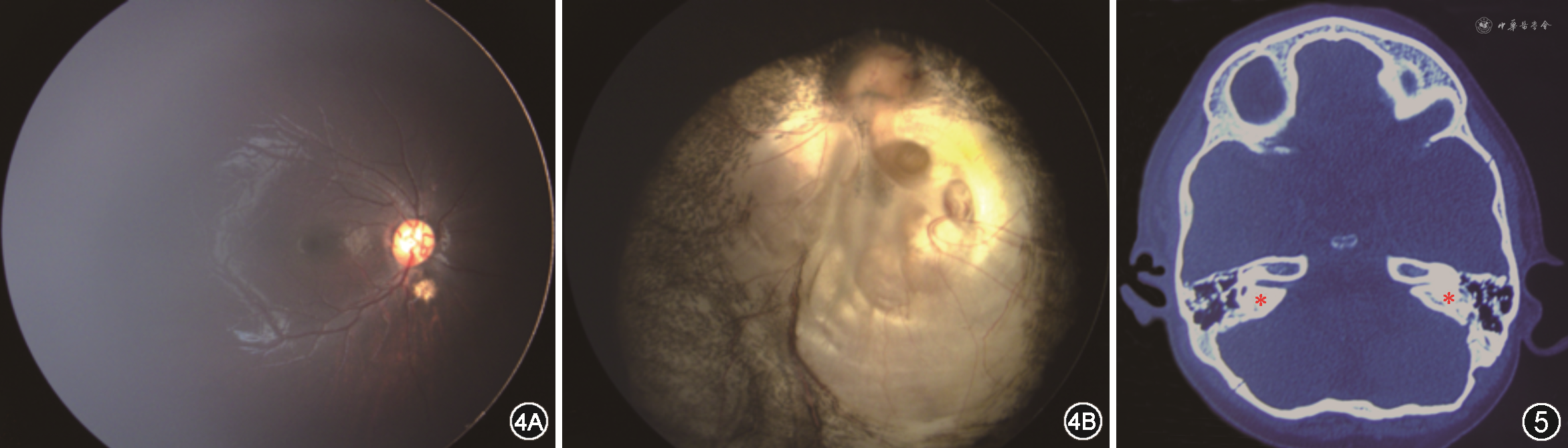
查体:患儿精神一般,行走不稳;面部不对称,左侧鼻唇沟变浅,额纹消失,口角向右侧偏斜(图1);右耳廓形态畸形,左耳廓结构畸形,部分缺损(图2,3),双侧外耳道未见明显异常;心前区未闻及病理性杂音。眼科检查:视力:条栅无明显注视行为,视动性眼震仪双眼阳性,单眼遮盖拒绝;眼位:33 cm映光交替遮盖:正位至+15°,偶有L/R 5°;遮盖去遮盖检查不配合;双眼各方位运动无明显受限,左眼睑闭合欠佳,双眼结膜无充血,角膜透明,瞳孔圆,直径约2.5 mm,直接、间接对光反射灵敏,晶状体透明。广角数码儿童视网膜成像系统(RetCam3,美国Clarity公司)眼底检查:右眼视盘边界清,色淡红,视网膜平伏,血管走行可,视盘下方见1/4 视盘直径大小脉络膜缺损灶(图4A);左眼视网膜平伏,下方大片状脉络膜缺损灶累及视盘及黄斑区(图4B)。1%阿托品散大瞳孔验光:右眼+1.75 DS(矫正不理解),左眼-9.00 DS(矫正不理解)。眼部B超:眼轴长右眼20.8 mm,左眼24 mm,左眼球后壁不光滑,双眼未见视网膜脱离光带。纤维鼻咽喉镜检查:双侧腺样体稍大,双侧扁桃体大,鼻部发育无异常。颞骨CT检查:左侧听小骨发育不良,左侧蜗神经闭塞,左侧内听道狭窄,右侧蜗神经孔稍窄,双侧半规管显示不良,提示发育不全(图5)。听力检查提示双耳感音神经性耳聋(左耳极重度)。心脏彩色超声检查:动脉导管结扎术后,血流正常。泌尿生殖系统B超检查未见明显异常。外周血行NGS基因检测结果显示:患儿CHD7基因发现c.3392T>C(编码区第3392号核苷酸由T变为C)的杂合核苷酸变异,该变异导致1131号氨基酸由亮氨酸(Leu)变为脯氨酸(Pro)。

结合患儿病史、临床表现及辅助检查诊断为:CHARGE综合征(双眼先天性脉络膜缺损、左眼睑闭合不全、左眼知觉性内斜视、双眼屈光参差、左侧面神经麻痹、先天性耳廓畸形、感音神经性耳聋、运动发育迟缓、精神发育迟缓、心脏动脉导管结扎术后)。
CHARGE综合征,也称Hall-Hittner综合征,是一种累及多种器官的罕见先天性发育异常,在新生儿中的发病率为1/12 000~1/8 500[1, 2]。1979年Hall和Hittner等分别描述了该病的临床特征[3, 4]。1981年Pagon以各种病变的英文首字母命名本病为CHARGE综合征,包括:眼组织缺损(Coloboma)、先天性心脏病(Heart disease)、后鼻孔闭锁(Atresia choanae)、生长发育迟滞(Retarded growth)、生殖器发育不全(Genital hypoplasia)以及耳部畸形或耳聋(Ear anomalies and deafness)等[5]。CHARGE综合征的典型畸形主要发生于胚胎发育的第4~9周,即视网膜发育、口咽膜破裂、心管形成、耳蜗形成的关键期,但具体发病机制尚不明确。90%~95%的确诊患者中会出现染色体8q12上的CHD7基因突变,这是目前已知唯一与CHARGE综合征相关的致病基因[6, 7, 8]。该基因属于染色质解旋酶DNA结合蛋白(chromodomain helicase DNA-binding protein,CHD)家族,在调节基因转录中发挥重要作用,可干扰神经嵴细胞的迁移和胚胎发生。胎儿发育早期CHD7基因在神经管、未分化的神经上皮和神经嵴起源的间质中高度表达,之后只在眼、耳及嗅觉系统等处表达,这也是CHARGE综合征常见的畸形部位[8]。
CHARGE综合征可表现为散发的常染色体显性疾病,多无家族史,97%的CHD7基因突变为新发突变[6, 7]。目前一般认为,CHARGE综合征诊断中的主要指标包括:(1)眼畸形;(2)后鼻孔闭锁;(3)半规管发育不良。次要指标包括:(1)颅神经及脑干病变;(2)下丘脑-垂体功能异常;(3)耳畸形及听力障碍;(4)纵隔器官发育不良;(5)智力低下。具备上述3个主要指标或2个主要指标+2个次要指标,即为典型CHARGE综合征;具备2个主要指标+1个次要指标,为部分型;具备2个主要指标或1个主要指标+2个次要指标则为非典型[6]。诊断时主要需与22q11.2缺失综合征和Kabuki综合征(歌舞伎综合征)重点鉴别,这两种综合征在临床表现上与CHARGE综合征多有重合,但通常都不符合CHARGE综合征的完整诊断标准,且两者的致病基因分别为22q11.21~22q11.23缺失和8号染色体区段重复dup8(p22p23)[6, 7, 8]。
本例患儿具备眼组织缺损和半规管发育不良2个主要指标,伴有颅神经病变(面神经麻痹)、耳畸形及听力障碍、纵隔器官发育不良(房间隔缺损、动脉导管未闭)及精神运动发育迟缓4个次要指标,并检测到CHD7基因突变。符合典型CHARGE综合征诊断标准。因患儿父母未行基因检测,结合CHD7基因致病特点,考虑父母无临床表现,推测患儿可能为该基因位点新发突变。本例患儿因眼部症状于眼科检查过程中发现眼部病变,结合全身异常诊断为CHARGE综合征,为眼科明确诊断的罕见全身多器官先天畸形。CHARGE综合征的主要眼部特征是眼部组织缺损,可发生在眼前段和后段,影响眼睑、虹膜、视网膜、脉络膜、视盘或黄斑,可伴有小眼球、小角膜、白内障和眼睑缺损,多累及双眼。最常见的是具有双侧不对称的视网膜脉络膜缺损,达79%~90%[9, 10, 11, 12]。患者的视力损伤取决于缺损的位置和性质,视力可接近正常或轻中度异常,严重者无光感[13]。其他眼部特征还包括屈光不正、斜视、弱视、眼睑下垂,面神经麻痹所致眼睑闭合不全及所致的暴露性角膜炎等。部分患者可继发视网膜脱离[14]。
尽管该病在儿科、心血管科、耳鼻喉科、整形科等均有报道,但于眼科就诊时明确该病者极少见,提醒眼科医师对CHARGE综合征提高认知,以及在临床先天性眼部缺损尤其是双眼不对称的脉络膜缺损患者诊治过程中,需警惕该综合征的可能性。CHARGE综合征由于合并多系统损伤和功能障碍,如精神、智力发育迟缓、视力损伤以及其他的严重畸形等,故需要眼科、儿科、耳鼻喉科、心血管科及康复科等多学科联合诊治。
所有作者均声明不存在利益冲突

























