
1例27岁双眼渐进性视力下降男性患者,表现为双眼黄斑区节细胞受损,全身伴四肢肌萎缩改变,完善线粒体全外显子基因检测,诊断为轴突型腓骨肌萎缩症2A2A型。该病临床尚无有效治疗方法,营养线粒体可能缓解临床症状。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者男性,27岁。因双眼先后渐进性视力下降8个月余,于2021年8月13日就诊于解放军总医院眼科医学部。无眼痛、眼球转动痛,无色觉、亮度异常和对比度下降,不伴恶心呕吐等。6岁时出现四肢远端肌萎缩。否认父母近亲结婚,姐妹均体健,无家族疾病史。
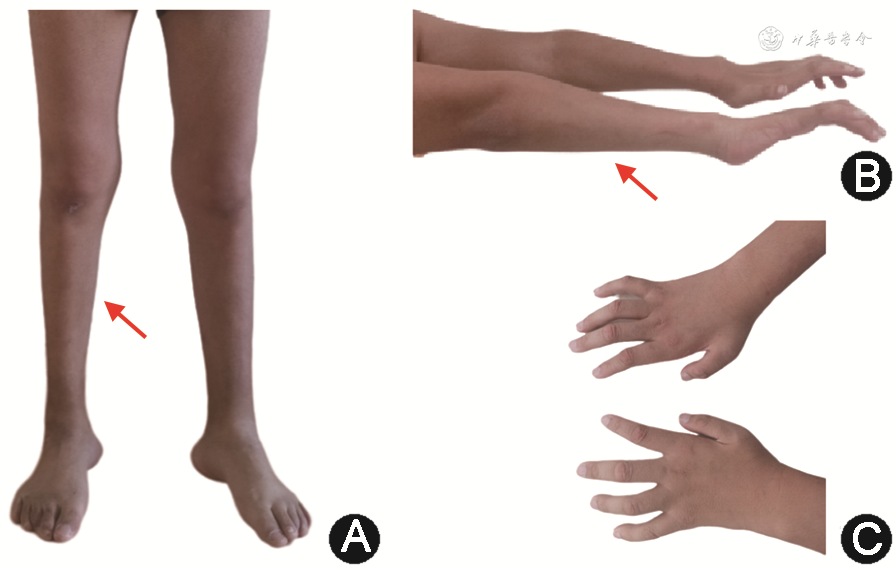
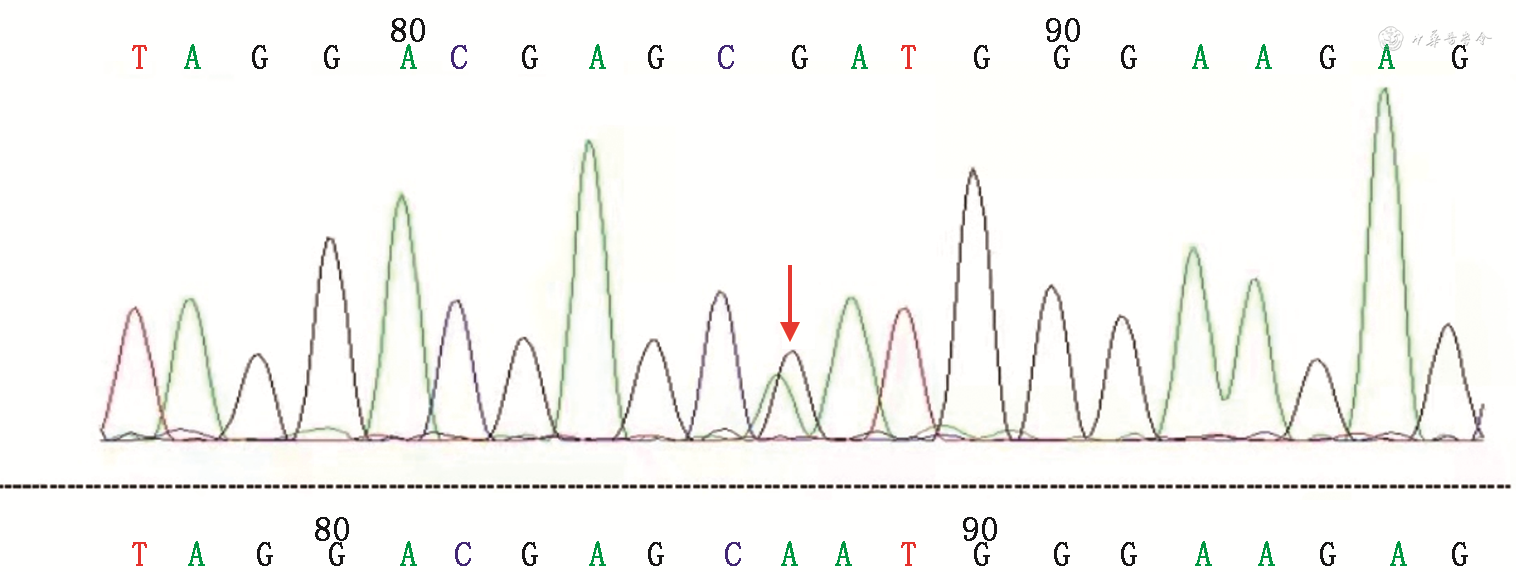
全身检查:颅神经未见明显异常。四肢活动远端稍受限,走路呈跨越步态。四肢肌肉萎缩,远端较为明显,肌容积明显减低(图1中A和B),双手畸形(图1中C),双踝关节强直。上肢肌力近端为3~4级,远端为2~3级;左下肢肌力近端为4+级,远端为1级;右下肢肌力近端为3级,远端为1级;腱反射(-)。患者无构音障碍、吞咽困难及认知障碍,粗试听力正常。实验室辅助检查(体液免疫、自身抗体谱、感染相关指标、生物化学指标等)未见明显异常。肌电图检查:正中神经传导速度减慢(29.1 ms),波幅降低(波幅差10.5%);四肢对称性多发性运动感觉性周围神经病,呈长度依赖性改变,以轴索病变为主继发脱髓鞘病变。全外显子组遗传疾病检测:MFN2基因杂合变异[c.319A>G(p.N107D),意义未明](图2),该位点基因突变来源于母亲,其母亲无症状。
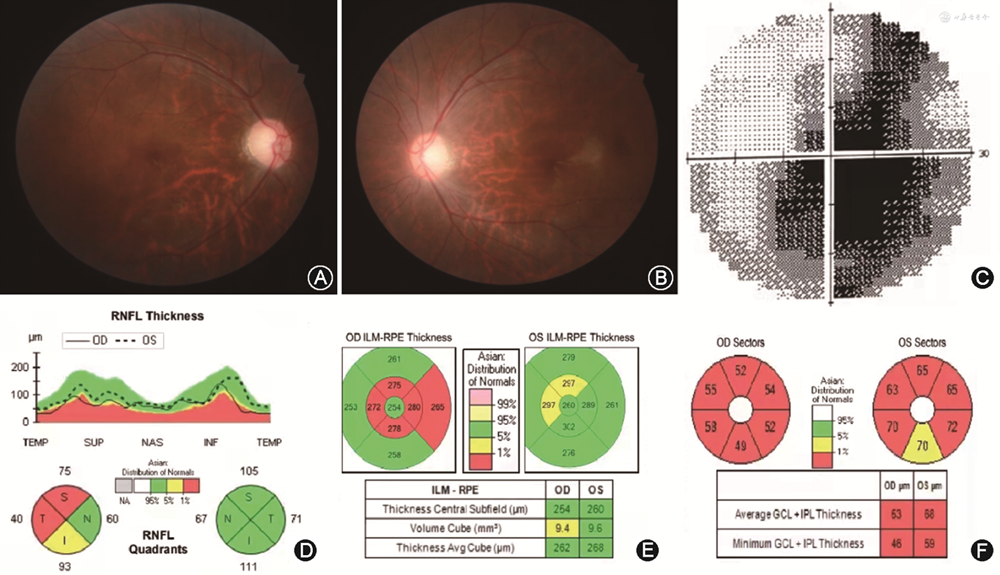
眼部检查:双眼最佳矫正视力均为0.01,眼压正常;相对性传入性瞳孔功能障碍(-),眼前节未见明显异常;双眼视盘界清、色淡(图3中A和B),余未见明显异常。视野检查:右眼中心偏下视野缺损(图3中C),左眼因固视差无法配合检查。相干光层析成像术(optical coherence tomography,OCT)检查:右眼视盘周围上方和颞侧视网膜神经纤维层(retinal nerve fiber layer,RNFL)变薄(图3中D),乳斑束受累(图3中E);双眼黄斑区节细胞层薄变(图3 中F)。闪光视觉诱发电位检查:双眼P2波潜伏期可,波幅降低。眼眶MRI检查(外院检查结果):双眼视神经均未见明显异常。
诊断:轴突型腓骨肌萎缩症(axonal charcot-marie-tooth disease,CMT)2A2A型;双眼视神经萎缩。给予营养线粒体治疗:口服艾地苯醌片90 mg/次,3次/日;辅酶Q10片10 mg/次,3次/日。
CMT2型中最为常见的基因型为CMT2A型,约占CMT2型的20%~30%。CMT2A2A型是由染色体1p36.2上MFN2基因杂合突变引起的常染色体显性遗传病,属于CMT2A型中的一种亚型,通常累及腓骨肌及其下肢远端肌肉,各年龄均可发病,发病年龄通常呈双峰分布,峰值在0~20和40~60岁[1]。该病发病越早,病情越严重。主要临床表现为高弓足,进行性远端肌无力,肌萎缩,跨越步态,远端感觉障碍,部分患者伴有视神经萎缩和听力障碍,家族内的疾病严重程度多相同,表明存在基因型表型相关性[2]。目前MFN2基因突变的病理机制尚不明确,普遍认为是线粒体动力学改变所致,MFN2基因突变通过改变线粒体融合和轴突微管系统的运输导致CMT2A型[3],线粒体融合和运输受损引起视神经萎缩。线粒体功能障碍可促进活性氧产生,从而损伤自身的动力学功能,加重临床表型,主要表现在能量需求较高的器官和组织[4]。2006年Züchner等[5]描述了CMT2A型家族6例亚急性病变患者,其中约60%患者伴有视神经萎缩;2012年Rouzier等[6]报道了1个在儿童早期发病的视神经萎缩家族,其与成年期轴突神经病变和线粒体肌病相关。2014年Bombelli等[7]报道了3例CMT2A型相关的视神经萎缩;2017年Ando等[1]分析了1 334例临床疑似CMT的日本患者,其中4例MFN2基因突变的CMT2A型患者伴视神经萎缩。2020年Guerriero等[3]报道了1例MFN2基因突变(c.2258duplT/p.Leu753fs)的CMT2A型患者,表现为双眼进行性视神经萎缩。
本例患者自幼四肢进行性远端肌无力、肌萎缩,成年后出现双眼进行性视神经萎缩,综合检查结果,诊断为CMT2A2A型。其基因突变来源于母亲,母亲尚未发病,患者的2个姐姐未进行基因检测,目前尚无相关临床症状。本病目前临床尚无有效治疗方法,营养线粒体可能缓解临床症状。MFN2基因突变通常导致青少年双眼视神经萎缩。本例患者为CMT2A2A型合并双眼视神经萎缩,OCT检查提示早期乳斑束受累,黄斑区节细胞受损较重,继发性引起右眼视盘RNFL薄变,符合线粒体肌病改变,而对侧眼尚未出现视盘RNFL薄变,可能因处于亚急性发作阶段。约60%亚急性发作CMT2型患者在几年内可出现视力、色觉和视野的缓慢恢复,但儿童时期出现的视神经萎缩往往不可逆[5]。因此,定期随访观察对于评估患者的视功能十分重要。在临床对于不明原因的视神经萎缩合并全身肌萎缩及步态异常者,应考虑本病,线粒体全外显子基因检测有助于诊断。
潘春艳, 白文浩, 孙明明, 等. MFN2基因突变致轴突型腓骨肌萎缩症2A2A型视神经萎缩1例[J]. 中华眼科杂志, 2023, 59(5): 408-410. DOI: 10.3760/cma.j.cn112142-20220611-00289.
所有作者声明无利益冲突

























