
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
2007年7月中华医学会心血管病学分会、中华心血管病杂志编辑委员会联合发布了我国"肺动脉高压筛查诊断与治疗专家共识"[1],该共识参考2003年在意大利威尼斯举办的第3次世界肺高血压大会对肺高血压的定义、临床分类、诊断流程和治疗策略,基于当时国际上临床实践的现状,并结合我国实际情况,对我国肺动脉高压(pulmonary arterial hypertension,PAH)的筛查、诊断与治疗均提出了切实可行的建议,对规范临床医生的诊疗行为、提高我国肺高血压临床诊治水平发挥了重要作用。
过去的11年间,国内外肺高血压领域研究进展迅速,新发现与PAH明确相关的基因,如CAV-1、KCNK3、TBX4、EIF2AK4、BMP9等[2,3,4,5,6],加深了对PAH这种复杂疾病发病机制的认识。2007年以前,全球范围内上市治疗PAH的药物只有静脉滴注依前列醇,皮下和静脉注射曲前列尼尔,吸入伊洛前列素,口服波生坦和西地那非片。2007年以后,吸入和口服曲前列尼尔,口服贝前列素、安立生坦、他达拉非、利奥西呱、马昔腾坦、司来帕格等药物陆续上市,PAH的治疗进入多元化阶段[7]。特别令人关注的是利奥西呱,被批准为全球第一个治疗慢性血栓栓塞性肺高血压(chronic thromboembolic pulmonary hypertension, CTEPH)的药物[8]。越来越多的医学中心开展了经皮肺动脉球囊扩张治疗CTEPH的临床研究,现阶段已发展为在光学相干断层成像术(optical coherence tomography, OCT)和肺动脉狭窄远端压力/近端压力(Pd/Pa)技术指导下的改良经皮肺动脉球囊成形术[9],使CTEPH的治疗由肺动脉内膜剥脱术单一治疗发展为外科、介入和药物治疗全面并进的新阶段,越来越多CTEPH患者的预后得到彻底改善。EARLY研究显示对世界卫生组织(World Health Organization, WHO)心功能Ⅱ级患者早期干预可明确改善预后[10],美国REVEAL注册登记研究首次建立PAH危险分层模型,建议根据危险分层制定治疗和随访策略[11],AMBITION研究显示安立生坦与他达拉非初始联合治疗可比单药治疗更好地改善患者预后[12],以及一些其他关键研究陆续发表,使PAH早期干预、危险分层以及初始联合治疗等策略逐渐得到公认。11年间,第4、5、6届世界肺高血压大会分别于2008、2013和2018年召开,对肺高血压的临床分类、发病机制、遗传、病理、危险分层、诊断方法和治疗标准与策略等进行了回顾、总结与归纳,不断更新认识。另外,2016年欧洲心脏病学会(ESC)和欧洲呼吸学会(ERS)也联合发布了欧洲新版肺高血压诊断和治疗指南[7]。
目前中国上市的药品有波生坦、安立生坦、马昔腾坦、他达拉非、吸入伊洛前列素、皮下和静脉注射曲前列尼尔及利奥西呱等。从2007年全国只有一两家肺血管疾病专科诊治中心,11年来陆续发展成为数十家肺高血压专科临床诊治中心,越来越多的心血管内科、呼吸内科、风湿免疫内科、心血管外科以及儿科医生投入到肺血管疾病的诊治工作中。
显然2007年版"肺动脉高压筛查诊断与治疗专家共识"已远不能支撑当前我国肺高血压领域的临床和研究工作。因此,中华医学会心血管病学分会肺血管病学组和中华心血管病杂志编辑委员会组织我国肺高血压领域的专家,全面总结国内外进展,特别是我国近11年来取得的成就,对2007年版共识进行了修订和更新。
本指南对推荐类别的表述沿用国际通用的方式。
Ⅰ类:指已证实和/或一致公认有益、有用和有效的操作或治疗。
Ⅱ类:指有用和/或有效的证据尚有矛盾或存在不同观点的操作或治疗。
Ⅱa类:有关证据/观点倾向于有用和/或有效,应用这些操作或治疗是合理的。
Ⅱb类:有关证据/观点尚不能被充分证明有用和/或有效,可考虑应用。
Ⅲ类:指已证实和/或一致公认无用和/或无效,并对一些病例可能有害的操作或治疗,不推荐使用。
对证据来源的水平表达如下。
证据水平A:资料来源于多项随机临床试验或荟萃分析。
证据水平B:资料来源于单项随机临床试验或多项非随机对照研究。
证据水平C:仅为专家共识意见和/或小型临床试验、回顾性研究或注册登记。
肺高血压是一类常见肺血管疾病[13],其主要病理生理学特征是静息状态下肺动脉压力升高,同时合并不同程度右心功能衰竭。
肺高血压指各种原因导致的肺动脉压力升高,包括毛细血管前性肺高血压、毛细血管后性肺高血压和混合性肺高血压(肺动脉和肺静脉压力均升高)。肺高血压的血流动力学诊断标准为:海平面状态下、静息时、右心导管测量肺动脉平均压(mean pulmonary artery pressure, mPAP)≥25 mmHg(1 mmHg=0.133 kPa)[7]。正常人mPAP为(14±3)mmHg,上限为20 mmHg。
PAH指孤立性肺动脉压力升高,而左心房与肺静脉压力正常,主要由肺小动脉本身病变导致肺血管阻力增加,且不合并慢性呼吸系统疾病、慢性血栓栓塞性疾病及其他未知因素等导致的肺高血压。PAH的血流动力学诊断标准为右心导管测量mPAP≥25 mmHg,同时肺小动脉楔压(pulmonary artery wedge pressure, PAWP)≤15 mmHg及肺血管阻力>3 Wood单位[7]。
特发性肺动脉高压(idiopathic pulmonary arterial hypertension, IPAH)是一类无明确原因、以肺血管阻力进行性升高为主要特征的恶性肺血管疾病。血流动力学符合PAH诊断标准。
关于上述术语的中英文翻译说明:肺高血压、肺动脉高压和特发性肺动脉高压特指英文专用术语"pulmonary hypertension"、"pulmonary arterial hypertension"和"idiopathic pulmonary arterial hypertension"的中文译称。学术界有人倾向将"pulmonary hypertension"、"pulmonary arterial hypertension"翻译为"肺动脉高压"和"动脉性肺动脉高压",但本指南仍保留2007年"肺动脉高压筛查诊断与治疗专家共识"所使用的术语:(1)参考我国学术界习惯用语"pulmonary embolism"一般都译为"肺栓塞"而不是常规译为"肺动脉栓塞",我国学术界基本公认"肺栓塞"就是肺动脉栓塞,几乎没有学者认为"肺栓塞"还包括"支气管动脉栓塞"或"支气管、肺泡内部堵塞"等引人异议的理解。(2)日本、台湾学者也均将"pulmonary hypertension"翻译为"肺高血压症"。
肺高血压血流动力学分类见表1。

肺高血压血流动力学分类
肺高血压血流动力学分类
| 定义 | 血流动力学特征 | 临床分型 | |
|---|---|---|---|
| 肺高血压 | mPAP≥25 mmHg | 所有五大类肺高血压 | |
| 毛细血管前性肺高血压 | mPAP≥25 mmHg,PAWP≤15 mmHg,PVR>3 Wood单位 | 肺动脉高压 | |
| 呼吸系统疾病和/或缺氧所致肺高血压 | |||
| 慢性肺动脉阻塞所致肺高血压 | |||
| 未知原因所致肺高血压 | |||
| 毛细血管后性肺高血压 | mPAP≥25 mmHg,PAWP>15 mmHg | 左心疾病所致肺高血压 | |
| 单纯毛细血管后性肺高血压 | DPG | 未知因素所致肺高血压 | |
| 混合性毛细血管后性肺高血压 | DPG≥7 mmHg和/或PVR>3 Wood单位 | ||
注:mPAP为肺动脉平均压,PAWP为肺小动脉楔压,PVR为肺血管阻力,DPG为肺动脉舒张压力阶差,dPAP为肺动脉舒张压;DPG=dPAP-PAWP;1 mmHg=0.133 kPa
本指南推荐的肺高血压临床分类仍延续既往五大类分类原则,并参照2018年第6届世界肺高血压大会最新内容进行修订,详见表2。
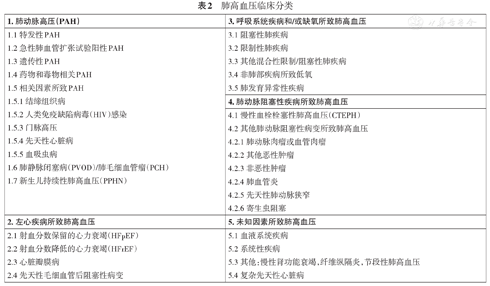
肺高血压临床分类
肺高血压临床分类
| 1.肺动脉高压(PAH) | 3.呼吸系统疾病和/或缺氧所致肺高血压 |
| 1.1特发性PAH | 3.1阻塞性肺疾病 |
| 1.2急性肺血管扩张试验阳性PAH | 3.2限制性肺疾病 |
| 1.3遗传性PAH | 3.3其他混合性限制/阻塞性肺疾病 |
| 1.4药物和毒物相关PAH | 3.4非肺部疾病所致低氧 |
| 1.5相关因素所致PAH | 3.5肺发育异常性疾病 |
| 1.5.1结缔组织病 | 4.肺动脉阻塞性疾病所致肺高血压 |
| 1.5.2人类免疫缺陷病毒(HIV)感染 | 4.1慢性血栓栓塞性肺高血压(CTEPH) |
| 1.5.3门脉高压 | 4.2其他肺动脉阻塞性病变所致肺高血压 |
| 1.5.4先天性心脏病 | 4.2.1肺动脉肉瘤或血管肉瘤 |
| 1.5.5血吸虫病 | 4.2.2其他恶性肿瘤 |
| 1.6肺静脉闭塞病(PVOD)/肺毛细血管瘤(PCH) | 4.2.3非恶性肿瘤 |
| 1.7新生儿持续性肺高血压(PPHN) | 4.2.4肺血管炎 |
| 4.2.5先天性肺动脉狭窄 | |
| 4.2.6寄生虫阻塞 | |
| 2.左心疾病所致肺高血压 | 5.未知因素所致肺高血压 |
| 2.1射血分数保留的心力衰竭(HFpEF) | 5.1血液系统疾病 |
| 2.2射血分数降低的心力衰竭(HFrEF) | 5.2系统性疾病 |
| 2.3心脏瓣膜病 | 5.3其他:慢性肾功能衰竭,纤维纵隔炎,节段性肺高血压 |
| 2.4先天性毛细血管后阻塞性病变 | 5.4复杂先天性心脏病 |
普通人群中肺高血压患病率约为1%,年龄>65岁人群中更是高达10%,以左心疾病所致肺高血压和呼吸系统疾病和/或缺氧所致肺高血压最为常见[14]。其中约80%患者来自发展中国家,基础疾病多为先天性心脏病和感染性疾病等。PAH发病率和患病率分别为5~10/百万人年和15~60/百万[15],约半数为IPAH、遗传性PAH或药物相关PAH,相关因素PAH则以结缔组织病最为常见,其中系统性硬化症约占结缔组织病相关PAH的2/3[16]。近年来IPAH平均诊断年龄为50~65岁,较20世纪80年代的36岁显著升高,原因尚不明确[17]。
在缺乏PAH靶向药物的传统治疗时代,美国原发性肺高血压的1、3和5年生存率分别为68%、48%和34%[18]。自20世纪90年代以来PAH靶向药物陆续上市,2006年法国注册登记提示新发IPAH、家族性PAH及阿米雷司相关PAH患者1年生存率为89.3%[16],2010年法国随访研究结果显示,新发IPAH、遗传性PAH及阿米雷司相关PAH患者的1、2和3年生存率分别达到89%、68%和55%[19]。
根据与PAH发生的相关程度和致病性,将危险因素分为确定致病及可能致病,详见表3[7]。

确定和可能导致肺动脉高压的药物和毒物
确定和可能导致肺动脉高压的药物和毒物
| 确定 | 可能 |
|---|---|
| 阿米雷司 | 可卡因 |
| 芬氟拉明 | 苯丙胺 |
| 右芬氟拉明 | 苯丙醇胺 |
| 甲基苯丙胺 | L-色氨酸 |
| 苯氟雷司 | 圣约翰草(贯叶连翘) |
| 达沙替尼 | 干扰素α、干扰素β |
| 毒性菜籽油 | 烷基化药物,如丝裂霉素C、环磷酰胺等 |
| 博舒替尼 | |
| 直接抗丙肝病毒药物 | |
| 来氟米特 | |
| 中药青黛 |
PAH的发生发展过程与肺血管结构和/或功能异常(即肺血管重构)密切相关。肺血管床内膜损伤、中层肥厚、外膜增殖/纤维化导致肺动脉管腔进行性狭窄、闭塞,肺血管阻力不断升高,进而导致右心功能衰竭甚至死亡。但PAH的发病机制尚未完全阐明。现认为,肺血管重构是遗传因素(基因突变)、表观遗传因素(DNA甲基化、组蛋白乙酰化、微小RNA等)以及环境因素(如低氧、氧化应激、机械剪切力、炎症、药物或毒物等)共同作用的结果[23]。多种血管活性分子[内皮素、血管紧张素Ⅱ、前列环素、一氧化氮(nitric oxide, NO)、一氧化碳、硫化氢及二氧化硫、雌激素等][24,25]、多种离子通道(钾离子通道、钙离子通道)、多条信号通路[MAPK通路、Rho/ROCK通路、PI3K/AKT通路、骨形态发生蛋白(BMP)/转化生长因子β(TGF-β)通路、核因子κB(NF-κB)通路和Notch通路][22,23,26,27]也在肺血管重构中发挥重要调节作用。
基因突变是部分PAH患者最根本的病因,基因检测可从分子水平确诊PAH。IPAH和遗传性PAH均为单基因常染色体显性遗传,目前已知9个致病基因——BMPR2、BMP9、ACVRL1、ENG、SMAD9、BMPR1B、TBX4、CAV1和KCNK3,可解释50%~80%的遗传性PAH和20%~50%的遗传性散发型IPAH患者的病因[7,28]。
BMPR2是最主要的遗传性PAH和IPAH致病基因。西方人群中70%~80%的遗传性PAH患者和20%~40%的IPAH患者携带BMPR2基因突变[29]。中国人群中BMPR2突变比例在遗传性PAH和IPAH分别为53%和15%[30]。BMPR2突变与先天性心脏病相关PAH亦紧密相关,先天性心脏病相关PAH患者中BMPR2基因突变率为7.5%,而先天性心脏病术后PAH患者中BMPR2突变率为12.3%[31]。BMPR2基因突变的外显率(即致病基因突变携带者最终发生PAH的比率)约为20%,且受性别影响,男性携带者外显率为14%,女性携带者外显率为42%,且基因突变与临床表型紧密相关[28]。与不携带突变的患者相比,携带BMPR2突变的IPAH/遗传性PAH患者发病更早,临床表型更重,预后更差[29,30]。
BMP9是最新发现的IPAH致病基因[6]。BMP9基因突变使IPAH发病风险上升22倍,可解释6.7%中国IPAH患者的遗传病因。BMP9突变的强烈致病性使其成为仅次于BMPR2排名第2的IPAH致病基因。
遗传性出血性毛细血管扩张症相关PAH为单基因常染色体显性遗传,其中ACVRL1和ENG是最主要的致病基因,可解释71%中国患者的病因[32]。
由于基因突变在PAH发生发展中发挥着重要作用,基因诊断对于患者临床诊断、治疗及患者家属的早期预警非常重要。应建立IPAH、遗传性PAH、遗传性出血性毛细血管扩张症相关PAH可疑患者的基因诊断程序,对有明确家族史的患者应绘制完整家系图谱,并对所有直系亲属进行临床检查。首先对家族中PAH先证者进行遗传检测,如发现明确的致病突变,则其直系亲属均应检测此致病突变,并对携带致病突变的亲属再次进行临床评估并长期随访。
PVOD和PCH为常染色体隐性遗传病,主要由EIF2AK4基因突变引起[5]。几乎全部的遗传性PVOD/PCH及9%~25%的散发患者携带EIF2AK4基因的纯合突变或复合杂合突变[33]。对于临床疑似PVOD/PCH,推荐进行遗传学检测。如检出EIF2AK4双等位基因突变,可从分子水平确诊PVOD/PCH。由于PVOD/PCH为常染色体隐性遗传病,需用Sanger一代测序在患者父母中检测致病突变,确认遗传模式。
PAH主要累及直径<500 μm的肺小动脉,特征性病理改变包括肺动脉中膜肥厚、内膜向心性或偏心性增殖和纤维化、外膜增厚纤维化、血管周围炎症细胞浸润及管腔内原位血栓形成等,严重患者可见复合病变,如丛样病变、扩张型病变等。左心疾病所致肺高血压则以肺静脉重构(静脉肌型化)为主,疾病晚期也可导致肺小动脉重构。PAH中结缔组织病相关PAH、遗传性出血性毛细血管扩张症相关PAH、PVOD/PCH、呼吸系统疾病和/或缺氧所致肺高血压、CTEPH及未知因素所致肺高血压则可能同时累及肺动脉和肺静脉,部分患者甚至合并肺毛细血管扩张或增殖性改变[34,35]。CTEPH病理改变包括栓塞肺动脉血栓机化、内膜增生导致管腔狭窄甚至闭塞,另外未发生栓塞的肺动脉亦可出现重构,部分肺毛细血管出现增殖样改变,肺静脉内膜纤维性增厚导致管腔狭窄,甚至支气管动脉迂曲扩张等[36]。
肺高血压临床表现差异很大,往往以活动后胸闷、气短及右心功能不全为主要特征。诊断分类复杂,建议临床医师接诊可疑肺高血压患者后及时转诊到肺血管疾病区域医疗中心进行诊断评价。危重患者不宜转诊时,应邀请肺血管疾病专家指导诊治。
推荐意见:临床医师应加强肺高血压诊断意识,对疑诊患者应详细询问肺高血压相关症状及诱发因素,收集既往疾病史、家族史、生育史、减肥药或毒物接触史及高原居住史等,并完善体格检查(Ⅰ,C)。
临床疑诊肺高血压的患者,应完善实验室检查[血常规、电解质、转氨酶、胆红素、肌酐、尿酸、D-二聚体、B型利钠肽(BNP)或N末端B型利钠肽原(NT-proBNP)、甲状腺功能及动脉血气分析等],综合评估病情(Ⅰ,C)。
临床疑诊肺高血压的患者,应常规进行心电图、胸部X线平片、超声心动图、呼吸功能、CT肺动脉造影、肺通气灌注显像、心肺运动试验等检查,尽早完善右心导管检查,进行准确诊断分类(Ⅰ,C)。
肺高血压早期没有特异性临床表现,绝大多数患者就诊时间明显延迟,至少1/5患者从症状出现至确诊时间超过2年。超过半数的IPAH患者确诊时WHO心功能为Ⅲ~Ⅳ级。部分肺高血压患者早期可能仅表现为基础疾病相关症状,当肺动脉压力明显升高时可出现右心功能衰竭症状。肺高血压最常见症状为活动后气促,其他症状包括乏力、头晕、胸痛、胸闷、心悸、黑矇、晕厥等。合并严重右心功能不全可出现下肢浮肿、腹胀、胃纳差、腹泻和肝区疼痛等。部分患者因肺动脉扩张引起机械压迫症状(如压迫左喉返神经引起声音嘶哑,压迫气道引起干咳,压迫左冠状动脉主干导致心绞痛等);肺动静脉畸形破裂或代偿扩张的支气管动脉破裂引起咯血。此外,还应询问患者是否具有可导致肺高血压基础疾病相关症状,如结缔组织病可出现雷诺现象、关节疼痛、口干、眼干、龋齿、脱发、皮肤硬化等。儿童患者还应格外注意发育情况,如发育明显异常或迟缓,则应重点筛查遗传代谢性疾病和内分泌疾病。
右心扩大可导致心前区隆起,肺动脉压力升高可出现肺动脉瓣第二心音亢进,三尖瓣关闭不全引起三尖瓣区收缩期杂音。严重右心功能不全时可出现颈静脉充盈或怒张、肝脏肿大、下肢水肿、多浆膜腔积液、黄疸和发绀等体征。右心室肥厚可导致剑突下抬举性搏动,出现第三心音表示右心室舒张充盈压升高及右心功能不全,约38%的患者可闻及右心室第四心音奔马律。
需要强调体格检查可能发现肺高血压潜在病因,如儿童及中青年患者或既往有先天性心脏病患者出现紫绀和杵状指(趾),往往提示艾森曼格综合征或复杂先天性心脏病;差异性紫绀和杵状趾是动脉导管未闭合并PAH的特征性表现;反复自发性鼻衄、体表皮肤毛细血管扩张提示遗传性出血性毛细血管扩张症;肩胛区收缩期血管杂音往往提示肺动脉阻塞性疾病,如大动脉炎或纤维纵隔炎累及肺动脉;肺野外围闻及血管杂音提示肺动静脉瘘可能;双肺吸气相爆裂音考虑肺间质疾病。
应重点询问有无先天性心脏病、结缔组织病、左心疾病、慢性肺部疾病、睡眠呼吸暂停、静脉血栓栓塞症,人类免疫缺陷病毒(HIV)感染、慢性肝病、血液系统疾病、甲状腺疾病、血吸虫感染和鼻衄病史等。儿童还需询问有无遗传代谢性疾病史。个人史需注意有无危险因素接触史如印刷厂和加油站工人接触油类物质、高原居住史、特殊用药史(食欲抑制剂类减肥药、达沙替尼、来氟米特和干扰素等)及吸毒史(甲基苯丙胺和可卡因)等。
需询问肺高血压患者有血缘关系的亲属中有无确诊或可疑肺高血压患者,有无反复鼻衄和皮肤毛细血管扩张史,有助于判断是否为遗传性PAH或遗传性出血性毛细血管扩张症。女性CTEPH患者要注意有无习惯性流产史,男性CTEPH患者要询问其母亲、姐妹等直系亲属有无习惯性流产等病史,有助于判断是否存在抗磷脂综合征。
心电图可为肺高血压提供诊断、鉴别诊断和预后判断的重要信息,但不能作为诊断或排除肺高血压的依据。肺高血压患者典型心电图表现为电轴右偏、右心房扩大和右心室肥厚征象,合并左心疾病时表现为双心房增大征象。Ⅰ导联S波振幅>0.21 mV诊断PAH的敏感度和特异度分别为89%和81%[37];QRS波增宽(≥120 ms)是IPAH预后不佳的预测因子[38]。肺高血压患者常合并心律失常,尤其是快速型房性心律失常,如阵发性房性心动过速、心房扑动(房扑)、心房颤动(房颤)等。PAH患者房扑和房颤5年累积发病率为25.4%,恢复窦律有助于改善病情[39]。PAH患者发生快速型室性心动过速相对少见。
PAH患者胸部X线平片常见征象有肺动脉段凸出及右下肺动脉扩张,伴外周肺血管稀疏(肺野透过度增加),右心房、室扩大。合并左心疾病的肺高血压患者可有不同程度肺淤血表现,合并严重肺部疾病(慢性阻塞性肺疾病、肺间质疾病、胸廓畸形、胸膜改变表现)的肺高血压则有相应基础疾病表现,合并近端肺动脉闭塞或单侧肺动脉缺如患者往往提示病变侧肺门影变小以及相应区域肺血明显减少或消失。但胸部X线平片正常并不能排除肺高血压。
超声心动图是临床上最常用的肺高血压筛查诊断及病情评价方法[40],主要从以下3方面进行评估:(1)判断肺高血压:通过三尖瓣反流峰速估测右心室收缩压。其他支持征象包括右心室/左心室基部内径比值>1、室间隔变平或左移(左心室偏心指数>1.1)、肺动脉内径>25 mm、下腔静脉内径>21 mm及吸气时塌陷率<50%、收缩末期右心房面积>18 cm2、右心室流出道多普勒加速时间<105 ms和/或收缩中期切迹以及舒张早期肺动脉瓣反流速度>2.2 m/s等。(2)发现心内结构、功能异常或血管畸形等:如先天性房、室水平分流或动脉导管未闭提示先天性心脏病相关PAH。左心瓣膜狭窄、左心室壁增厚、左心室收缩/舒张功能异常提示左心疾病所致肺高血压。短期内大量心包积液需考虑结缔组织病相关PAH。肺动脉管腔内占位提示第四大类肺高血压可能。(3)右心功能评估:二维超声心动图无法直接评估右心功能,但可通过右心房大小、三尖瓣环收缩期位移(TAPSE)、Tei指数以及有无心包积液等间接评价,三维、四维超声心动图可提供更可靠的右心室容量和收缩功能测定结果[41,42]。
超声心动图诊断肺高血压的敏感度和准确度整体良好,但对于部分患者肺动脉压力估测值误差较大,因此临床评估时,应结合三尖瓣结构、三尖瓣反流信号强弱及其他支持征象来综合评估。如三尖瓣反流峰速难以测量时,肺动脉压力易被低估;当存在三尖瓣大量反流,尤其单纯右心疾病或三尖瓣疾病时,肺动脉压力易被高估。值得强调的是,只有在排除右心室流出道梗阻的前提下,三尖瓣反流峰速估测的右心室收缩压加上右心房压才近似于肺动脉收缩压。
推荐意见:超声心动图检查提示三尖瓣峰反流速度≤2.8 m/s,无其他肺高血压征象,则肺高血压可能性低,建议定期复查超声心动图(Ⅱa,C)。
超声心动图检查提示三尖瓣峰反流速度≤2.8 m/s,有其他肺高血压征象,或三尖瓣峰反流速度2.9~3.3 m/s,无其他肺高血压征象,则肺高血压中度可能,建议进一步检查包括右心导管检查(Ⅱa,C)。
超声心动图检查提示三尖瓣峰反流速度2.9~3.3 m/s,有其他肺高血压征象,或三尖瓣峰反流速度≥3.4 m/s,无论有无其他肺高血压征象,则肺高血压高度可能,建议进一步检查包括右心导管检查(Ⅰ,C)。
呼吸功能检查有助于发现潜在的肺实质或气道疾病。PAH患者通常有轻至中度外周小气道功能障碍,大部分患者弥散功能轻、中度下降[43,44]。IPAH患者如一氧化碳弥散量(DLco)显著降低(<45%预测值)往往提示心输出量明显降低,预示预后不良[45]。此外,合并肺间质疾病、PVOD/PCH以及部分IPAH患者(尤其是老年且有大量吸烟史)DLco也会显著降低。肺容积减少往往提示存在胸廓畸形、胸膜增厚或肺间质纤维化等疾病。
轻症PAH患者动脉血气分析结果可完全正常,病情严重时由于过度通气可表现为低碳酸血症和轻度低氧血症,如合并严重低氧血症应考虑存在动静脉分流可能,如先天性心脏病或肺动静脉畸形。IPAH患者动脉血二氧化碳分压越低提示代偿性过度通气越严重,预后越差,氧分压和预后无明确相关性[46]。部分肺高血压患者动脉血气分析可表现为氧分压降低而二氧化碳分压升高,提示存在气道梗阻,可由中枢性(脊髓空洞症、睡眠呼吸暂停、神经肌肉系统疾病)和气道本身疾病(慢性阻塞性肺疾病等)所致。
肺通气灌注显像是筛查CTEPH的重要手段。相比CT肺动脉造影,肺通气灌注显像敏感度更高[47]。部分PAH和PVOD/PCH也可出现小的外周肺野节段性灌注缺失。因此,肺通气灌注显像阴性可排除第四大类肺高血压,但阳性结果则不具特异性,需结合其他临床征象和影像学检查进一步明确诊断。
胸部CT可提供关于心脏、血管、肺实质及纵隔病变的详细信息,主要用于肺高血压病因诊断、肺血管介入影像学评估以及评价预后[48]。CT平扫发现以下征象提示肺高血压可能:肺动脉直径≥29 mm;主肺动脉直径/升主动脉直径比值≥1.0;大于3~4个亚段的肺动脉直径/支气管直径比值>1。高分辨率CT也可为诊断肺实质、肺间质疾病和PVOD/PCH提供重要依据。PVOD患者CT影像特征包括:肺间质水肿征象,弥漫小叶中心性磨玻璃影以及小叶间隔增厚,部分患者可合并纵隔淋巴结肿大。而PCH患者CT影像则往往仅存在弥漫小叶中心性磨玻璃影,一般不合并小叶间隔增厚和纵隔淋巴结肿大。需强调,临床上约1/3的PAH患者存在不同程度肺磨玻璃影征象,因此不能仅根据此征象进行诊断。
CT肺动脉造影是诊断肺血管畸形(肺动静脉瘘、肺动脉瘤、肺动脉夹层)和肺动/静脉阻塞性疾病(急性肺栓塞、CTEPH、大动脉炎、肺动脉肿瘤、纤维纵隔炎、肺静脉狭窄等)的关键技术手段之一。但对于外周肺动脉狭窄病变如外周型CTEPH或多发外周肺动脉狭窄患者,单纯进行CT肺动脉造影检查易漏诊。尽管CT影像技术不断进步,如双能CT已开始在临床应用[49],但仍有漏诊外周段或亚段水平肺动脉狭窄的可能,需结合肺通气灌注显像或直接肺动脉造影进行诊断。在有经验的中心CT肺动脉造影重建影像可部分替代肺动脉造影,用于指导肺动脉内膜剥脱术或肺动脉介入治疗的术前评估。此外,心脏结构CT可准确评估肺高血压患者是否合并先天性心脏病,尤其对那些易被超声心动图漏诊的先天性心脏病类型,如特殊部位房间隔缺损(上腔静脉、下腔静脉或冠状静脉窦型)、部分型肺静脉异位引流和双向分流动脉导管未闭等。
心脏磁共振是目前评价右心大小、形态和功能的"金标准",且具有较高的可重复性[7]。心脏磁共振可无创评估血流动力学状态、估测每搏量、心输出量、肺动脉弹性和右心室质量[50]。对疑诊肺高血压患者,心脏磁共振支持性诊断征象包括:右心扩大、肺动脉增宽、钆对比剂延迟强化、肺动脉弹性减低及肺动脉瓣反流等。心脏磁共振也可用于评价肺高血压患者病情严重程度及治疗效果。对于IPAH患者,心脏磁共振测定右心室射血分数变化对患者预后的预测价值显著优于心导管测定的肺血管阻力变化。因此,推荐对肺高血压患者进行心脏磁共振评估。对于超声心动图无法确诊的先天性心脏病患者,心脏磁共振也可提供更多诊断信息。对于疑诊肺动脉阻塞性疾病患者,注射对比剂或无对比剂磁共振肺动脉造影都具有一定诊断价值,尤其适合一些临床特殊情况,如孕妇、肾功能不全或对含碘对比剂过敏患者。
夜间低氧血症和阻塞性睡眠呼吸暂停是导致肺高血压的重要因素。对有可疑睡眠呼吸暂停症状、存在不明原因二氧化碳潴留的患者以及合并唐氏综合征的先天性心脏病患儿,应常规进行睡眠呼吸监测检查[51]。
目前血液学检查主要用于肺高血压病因筛查及判定器官损害情况。血细胞检查异常需警惕结缔组织病、血液系统疾病(尤其各种贫血、继发脾功能亢进、多发性骨髓瘤和骨髓异常增生综合征等)、慢性缺氧性疾病(红细胞数量、血红蛋白和红细胞比积显著升高)和其他系统性疾病。肝功能异常需考虑肝脏疾病、门脉疾病、血液系统疾病、心力衰竭或药物不良反应(如内皮素受体拮抗剂可导致转氨酶和胆红素水平升高)等情况。甲状腺功能、风湿免疫抗体、肝炎及艾滋病毒抗体也应作为肺高血压患者常规检查项目。对于病因不明的儿童肺高血压患者,推荐进行同型半胱氨酸和血、尿有机酸代谢检测,用于排除甲基丙二酸尿症。对于CTEPH患者应常规进行遗传性易栓症和获得性易栓症筛查,包括蛋白S、蛋白C和抗凝血酶Ⅲ活性检测,以及抗磷脂抗体、狼疮抗凝物、同型半胱氨酸和肿瘤标志物检测。所有肺高血压患者均推荐在基线评估和后续随访过程中进行NT-proBNP或BNP检测,用于评估病情并指导治疗。
腹部超声主要用于肺高血压病因筛查和病情严重程度评估。肺高血压患者如合并右心衰竭可出现严重肝、脾淤血,肝静脉扩张,腹腔积液等征象。对所有拟诊IPAH患者,尤其存在心输出量偏高时,应除外门脉高压(可继发于基础肝脏疾病或其他先天性疾病,如门脉海绵样变性等)或先天性肝外门体分流(Abernethy畸形)等情况。对于存在明显鼻衄及家族史患者,应通过腹部超声筛查是否存在肝脏动静脉瘘,疑诊遗传性出血性毛细血管扩张症时也应行肝脏增强CT和头颅磁共振进一步检查。
肺高血压是一个血流动力学概念,准确获取血流动力学数据并正确解读至关重要。右心导管检查是确诊肺高血压的"金标准",也是进行鉴别诊断、评估病情和治疗效果的重要手段。在有经验的中心,肺高血压患者进行右心导管检查安全性良好,并发症发生率仅为1.1%,死亡率仅为0.055%[52]。
零点校准:仰卧位时胸前壁和床面中间的位置作为零点校准位,代表左心房所在水平。
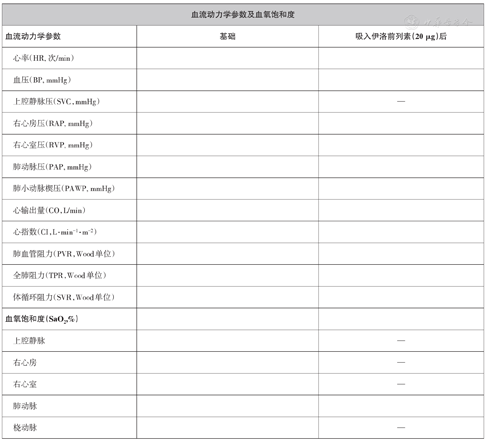
血流动力学参数的测定:推荐右心导管检查常规测定以下血流动力学参数:(1)心率、体循环血压(有创或无创压);(2)右心房压、右心室收缩压和舒张末压;(3)肺动脉收缩压、舒张压和平均压;(4)PAWP,如无法测定可应用左心室舒张末压(left ventricular end-diastolic pressure,LVEDP)参照,房间隔缺损患者可直接测量肺静脉压;(5)心输出量。需强调所有压力测量应在正常呼气末(非屏气状态)时测定。对无心内或大动脉分流患者,建议采用热稀释法测量心输出量,因为即使在低心输出量状态和/或严重三尖瓣反流患者中,该方法也可准确反映患者的实际心输出量。一般至少测定3次取均值。而对分流性先天性心脏病应采用Fick法测定肺循环(Qp)和体循环血流量(Qs)。Fick法分为直接法(通过设备直接测定氧摄取量和氧耗量)和间接法(估测氧耗量=体表面积×125 ml/min或通过量表估测氧耗量)。国内均采用间接Fick法测定。
血氧指标:(1)腔静脉、右心房、右心室及肺动脉氧饱和度。对于无心内或大血管分流患者,从右心房到肺动脉,各腔室氧饱和度依次递减或改变不明显。(2)若以上不同部位血氧饱和度明显异常,应进一步检查确诊。(3)混合静脉氧饱和度(SvO2),对于无心内或大血管分流患者,SvO2指肺动脉近端血氧饱和度。(4)对于房间隔缺损患者应尽量测定肺静脉氧饱和度。
导管和操作路径选择:漂浮导管可测量PAWP,并配合相应功能组件进行热稀释法心输出量测定,故为肺高血压患者行右心导管检查首选。对肺高血压患者进行右心导管检查最常用的径路为右颈内静脉、股静脉和左前臂静脉[53]。如无特殊必要,漂浮导管不推荐股静脉径路。
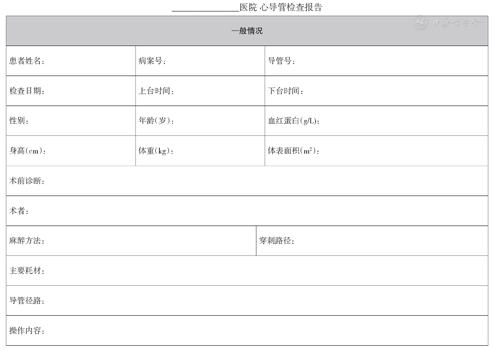
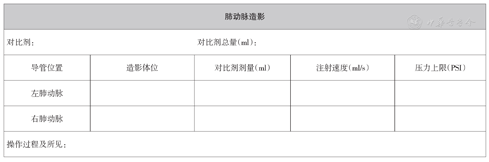
右心导管检查模板见附件1。
推荐意见:右心导管检查是确诊肺高血压、诊断分类及治疗指导的重要检查手段(Ⅰ,C)。
建议患者转诊到肺血管疾病区域医疗中心进行右心导管检查,检查结果更全面,安全性更高(Ⅰ,B)。
评估肺高血压疗效可进行右心导管检查(Ⅱa,C)。
评估先天性心脏病是否适合介入封堵或手术修补需进行右心导管检查(Ⅰ,C)。
左心疾病或呼吸系统疾病所致肺高血压拟行器官移植前需进行右心导管检查(Ⅰ,C)。
右心导管检查无法获得PAWP,可以测量的左心室舒张末压代替(Ⅱa,C)。
疑诊左心疾病或呼吸系统疾病所致肺高血压时应进行右心导管检查,以明确诊断和指导治疗(Ⅱb,C)。
CTEPH患者应进行右心导管检查明确诊断及指导治疗(Ⅰ,C)。
急性肺血管扩张试验:少数PAH由肺动脉痉挛引起,单独应用大剂量钙通道阻滞剂可显著改善症状、血流动力学和长期预后[54]。急性肺血管扩张试验是筛选此类患者的有效方法。尽管其他PAH亚类中也有少数患者符合急性肺血管扩张试验阳性标准,但难以从单纯钙通道阻滞剂治疗中持续获益[55]。故仅推荐对于IPAH、遗传性PAH和药物相关PAH患者首次右心导管检查时行急性肺血管扩张试验。
急性肺血管扩张试验药物与方法:用于试验的药物均为起效迅速、半衰期短的选择性肺血管扩张药物,包括吸入伊洛前列素[56]、静脉泵入腺苷[57]、吸入NO或静脉泵入依前列醇。目前国内主要应用吸入用伊洛前列素(商品名:万他维)和腺苷(商品名:艾朵)进行检查。具体使用方法见表4。

急性肺血管扩张试验药物使用方法
急性肺血管扩张试验药物使用方法
| 药物 | 给药途径 | 半衰期 | 剂量范围 | 使用方法 |
|---|---|---|---|---|
| 伊洛前列素 | 雾化吸入 | 5~25 min | 10~20 μg(指装入雾化吸入装置剂量) | 推荐空气压缩式雾化器,保证雾化颗粒大小适合沉积于肺泡,直接应用伊洛前列素原液或进行1∶1稀释后使用,吸入5~10 min,观察10~15 min,测定肺动脉压力下降幅度最大值 |
| 腺苷 | 静脉泵入 | 50~200 μg·kg-1·min-1 | 50 μg·kg-1·min-1起始泵入,每隔2 min上调25 μg·kg-1·min-1,直至阳性或出现不能耐受的不良反应或最大剂量(200 μg·kg-1·min-1) | |
| 一氧化氮 | 吸入 | 15~30 s | 10~20 p.p.m. | 吸入5 min |
| 依前列醇 | 静脉泵入 | 3 min | 2~12 ng·kg-1·min-1 | 2 ng·kg-1·min-1起始泵入,每隔10 min上调2 ng·kg-1·min-1 |
急性肺血管扩张试验阳性标准[7]:mPAP下降幅度超过10 mmHg且绝对值≤40 mmHg,同时心输出量增加或不变,需同时满足以上3项标准方为阳性。仅有不足10%的IPAH患者急性肺血管扩张试验阳性,可接受大剂量钙通道阻滞剂治疗(详见治疗中的钙通道阻滞剂部分)。服药1年后应再次复查右心导管和急性肺血管扩张试验,仅半数阳性患者表现为持续阳性,可继续单用钙通道阻滞剂治疗;而转阴患者建议给予PAH靶向药物治疗。
推荐意见:建议到肺血管疾病区域医疗中心进行急性肺血管扩张试验检查(Ⅰ,C)。
建议对IPAH、遗传性PAH及药物相关PAH患者常规进行急性肺血管扩张试验,以筛查适合大剂量钙通道阻滞剂治疗的患者(Ⅰ,C)。
推荐雾化吸入伊洛前列素或静脉泵入腺苷进行急性肺血管扩张试验(Ⅰ,C)。
不建议采用吸氧、静脉注射钙通道阻滞剂进行急性肺血管扩张试验(Ⅲ,C)。
不建议对除IPAH、遗传性PAH及药物相关PAH以外的PAH或其他类型肺高血压患者进行急性肺血管扩张试验(Ⅲ,C)。
肺动脉造影:肺动脉造影是评价肺血管形态及血流分布的重要手段,可结合CT肺动脉造影、肺通气灌注显像等其他影像技术对肺血管畸形或肺动脉/静脉狭窄性疾病进行诊断。为清晰显示双侧肺动脉远端分支及肺动脉血流情况,对高度疑诊肺血管畸形或狭窄患者建议分别行双侧肺动脉造影,而对于段或亚段一级肺动脉病变为主的患者,则需进行超选择肺动脉造影。造影模式一般首选数字减影造影,能更好显示肺动脉外周灌注情况。但对于病情严重或其他原因无法配合屏气患者可直接按心脏造影模式进行。为更充分暴露病变部位,对于两侧肺动脉近端病变,首选同侧造影,即右肺动脉近端病变选择右前斜位(30°~60°),左肺动脉近端病变选择左前斜位(30°~60°);而对于双下肺动脉远端病变,则首选对侧造影,即右肺动脉选择左前斜位(30°~60°),左肺动脉则选择右前斜位(30°~60°)。如单一体位无法充分暴露目标血管,则需多体位投照。
肺动脉造影潜在风险包括对比剂过敏、对比剂肾病、右心衰竭加重、肺动脉高压危象甚至猝死[58]。因此肺动脉造影应尽量选择等渗对比剂,对于血流动力学不稳定的患者行肺动脉造影应谨慎,造影时应适当减缓注射流速及减少剂量。
肺动脉腔内影像技术:目前可采用的肺动脉腔内影像技术包括血管内超声(intravenous ultrasound, IVUS)和光学相干断层成像术(optical coherence tomography, OCT),主要用于肺动脉腔内和管壁形态学评估和血管功能评价,现阶段主要用于慢性闭塞性肺血管病的鉴别诊断和指导介入治疗,在PAH患者病情评估和预后评价方面也有一定价值[59,60,61,62,63]。两种技术各有特点,OCT成像清晰度更高,但景深较小,适合评价5 mm以下肺动脉腔内形态。IVUS清晰度虽然不足,但景深较大,更适合评价肺动脉血流功能及肺动脉机械性特征(包括血管顺应性、扩张性和弹性模量等)。CTEPH患者行OCT检查可在狭窄血管内发现不同程度的内膜分隔影像,而且在肺动脉造影所见狭窄中几乎均可发现上述征象。而对于IPAH或结缔组织病相关PAH或大动脉炎累及肺动脉造成肺动脉狭窄患者,IVUS或OCT影像主要表现为管壁内膜向心性增厚导致管腔面积减少。PAH患者的IVUS和OCT影像均提示肺动脉管壁显著增厚,而且增厚程度与肺动脉压和肺血管阻力呈正相关,而肺动脉机械性能降低则与肺动脉压和肺血管阻力呈负相关。此外,在结缔组织病相关PAH患者中,IVUS影像提示<5 mm远端分支血管重构的患者预后要明显劣于仅提示>5 mm近端肺动脉分支重构的患者[59]。
建议对疑诊肺高血压的患者首先考虑常见疾病如第二大类的左心疾病和第三大类的呼吸系统疾病,然后考虑CTEPH,最后考虑PAH和未知因素所致。对疑诊PAH的患者应考虑相关疾病和/或危险因素导致的可能,仔细查找有无家族史、先天性心脏病、结缔组织病、HIV感染、门脉高压、与肺动脉高压有关的药物服用史和毒物接触史等。肺高血压的诊断流程见图1。
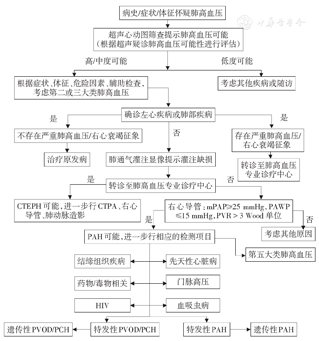
CTEPH:慢性血栓栓塞性肺高血压,CTPA:CT肺动脉造影,mPAP:肺动脉平均压,PAWP:肺小动脉楔压,PVR:肺血管阻力,PAH:肺动脉高压,HIV:人类免疫缺陷病毒,PVOD:肺静脉闭塞病,PCH:肺毛细血管瘤;1 mmHg=0.133 kPa
推荐意见:超声心动图是疑诊肺高血压时一线无创诊断方法(Ⅰ,C)。
无法解释肺高血压需排除CTEPH时建议进行肺灌注或肺通气灌注显像检查(Ⅰ,C)。
疑诊CTEPH时建议进行CT肺动脉造影检查(Ⅰ,C)。
所有肺高血压患者均应进行血常规、血生化、自身免疫抗体、HIV抗体、甲状腺功能检查(Ⅰ,C)。
PAH考虑门脉高压引起时建议进行腹部超声检查(Ⅰ,C)。
肺高血压首次评估时建议完善呼吸功能包括DLco检查(Ⅰ,C)。
所有肺高血压患者均应完善高分辨肺CT检查(Ⅱa,C)。
CTEPH患者应进行肺动脉造影检查(Ⅱa,C)。
不建议对肺高血压患者进行开胸或胸腔镜肺活检(Ⅲ,C)。
目前推荐使用1998年制定的WHO心功能分级评价肺高血压患者心功能状态,与纽约心脏协会心功能分级相比,强调晕厥症状的重要性,是评估病情严重程度及预后的重要指标。未治疗的IPAH和遗传性PAH平均生存时间与WHO心功能分级密切相关,Ⅳ级者为6个月,Ⅲ级者为2.5年,Ⅰ~Ⅱ级者为6年[64]。
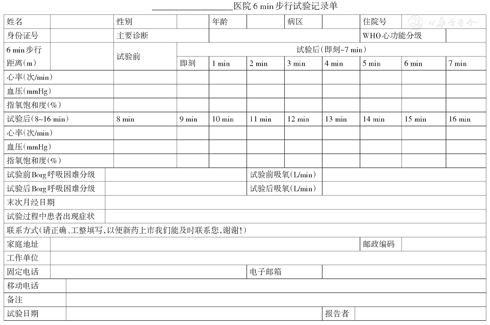
6MWT是一种客观评价患者运动耐量的方法,具有设备要求简单、经济、重复性好及便于规范化操作的优点[65,66]。首次住院的患者6 min步行距离与预后明显相关,而6MWT后1 min心率恢复绝对值也已成为预测预后的重要指标[67]。建议肺高血压患者首诊时均进行6MWT,并定期复查。Borg呼吸困难分级指数与6 min步行距离结合可评价肺高血压患者的心肺功能及尽力程度。需强调,6 min步行距离可能受患者身高、体重、性别、年龄、合并疾病、性格及情绪等因素影响,在临床解读时要充分考虑。6MWT记录单和Borg呼吸困难分级见附件2、3。
心肺运动试验是一项从静息到运动整体定量评估心肺功能的重要检查方法。心肺运动试验测定方法在肺高血压人群中尚未统一,大多数中心使用递增斜坡方案[68]。越来越多的证据支持心肺运动试验用于评价肺高血压患者运动功能受损、药物疗效及预后。PAH患者运动耐量、有氧代谢能力和通气效率明显受损,可表现为呼气末二氧化碳分压(PCO2)降低,二氧化碳通气量(VE/VCO2)升高,氧脉搏(VO2/HR)和峰值氧摄取量(PVO2)降低[69]。PAH患者VE/VCO2斜率≥45、最大摄氧量(VO2max)<10.4 ml·min-1·kg-1、呼气末二氧化碳分压(PETCO2)<20 mmHg则预示PAH患者临床恶化事件发生率明显升高,需要更加积极的药物干预[70,71]。
尽管肺高血压诊断意识已明显提高,但大多数患者确诊时仍为疾病晚期。临床医师应对肺高血压高危人群定期进行超声心动图筛查,以便早期诊断、早期治疗。尤其应定期对PAH相关基因(BMPR2或其他相关基因)突变携带者、IPAH及遗传性PAH患者亲属、结缔组织病患者(尤其是系统性硬化症患者)、先天性心脏病患者、门脉高压患者、HIV感染者和静脉血栓栓塞症患者等进行超声心动图检查。
因目前尚无单独指标能准确判断患者病情和评估预后,故需综合多个临床指标进行评估。第6届世界肺高压大会推荐使用简化的危险分层量表(表5),通过评估治疗前基础状态和短期治疗(3~6个月)后的关键临床指标来预测患者长期预后[72]。需强调,目前推荐的危险分层量表仅适用于成人PAH患者。其他类型肺高血压和儿童PAH尚缺乏统一的危险分层量表。
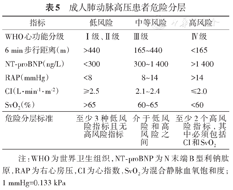
成人肺动脉高压患者危险分层
成人肺动脉高压患者危险分层
| 指标 | 低风险 | 中等风险 | 高风险 |
|---|---|---|---|
| WHO心功能分级 | Ⅰ级、Ⅱ级 | Ⅲ级 | Ⅳ级 |
| 6 min步行距离(m) | >440 | 165~440 | <165 |
| NT-proBNP(ng/L) | <300 | 300~1 400 | >1 400 |
| RAP(mmHg) | <8 | 8~14 | >14 |
| CI(L·min-1·m-2) | ≥2.5 | 2.1~2.4 | ≤2.0 |
| SvO2(%) | >65 | 60~65 | <60 |
| 危险分层标准 | 至少3种低风险指标且无高风险指标 | 介于低风险和高风险之间 | 至少2个高风险指标,其中必须包括CI和SvO2 |
注:WHO为世界卫生组织,NT-proBNP为N末端B型利钠肽原,RAP为右心房压,CI为心指数,SvO2为混合静脉血氧饱和度;1 mmHg=0.133 kPa
推荐意见:建议对PAH患者病情严重程度进行评估,包括心功能、运动耐量、血清生化标记物、超声心动图及血流动力学指标等(Ⅰ,C)。
病情稳定的PAH患者建议每3~6个月随访1次(Ⅰ,C)。
建议给予充分靶向药物治疗,使PAH患者病情达到或维持低危状态(Ⅰ,C)。
若PAH患者经靶向药物治疗病情仍进展或维持中危状态,应考虑靶向药物治疗不充分(Ⅱa,C)。
肺高血压患者妊娠期病死率显著升高,生育期女性患者应严格避孕。尽管急性肺血管扩张试验阳性IPAH患者的妊娠安全性已有明显改善[73],但仍应在肺血管疾病专科和产科医师的严密随访下进行,剖宫产为此类患者首选方案。
病情相对稳定的患者应进行适度运动和康复训练,有助于提高运动耐量、心肺功能和改善生活质量[74]。建议在有经验的心脏或呼吸病中心接受康复训练,运动以不引起明显气短、眩晕、胸痛为宜。
择期手术会增加肺高血压患者病情恶化的风险,应尽可能采用局部或区域阻滞麻醉,避免全身麻醉,尤其是需气管插管的全身麻醉手术。
感染可导致肺高血压患者病情加重,推荐在秋冬交替季节接种流感疫苗和肺炎链球菌疫苗,降低肺部感染发生风险。
肺高血压患者易产生不同程度的焦虑和/或抑郁状态,应充分考虑并评估患者的精神心理状态,鼓励家属给予心理支持,必要时请专科医师进行干预和疏导。
约1/4的肺高血压患者在飞行过程中会出现低氧状态(定义为指尖氧饱和度<85%)。因此WHO心功能较差(Ⅲ、Ⅳ级)或动脉血氧分压<60 mmHg时需谨慎飞行或在飞行过程中有氧气支持。此外,肺高血压患者应避免前往高海拔(1 500~2 000 m以上)地区或低氧环境。
CTEPH患者需终生抗凝,IPAH、遗传性PAH和减肥药相关PAH如无抗凝禁忌证可考虑长期抗凝治疗[75],而其他类型肺高血压尚无证据支持抗凝治疗可使患者获益。但合并矛盾性栓塞的艾森曼格综合征以及合并肺动脉原位血栓形成的患者需酌情抗凝治疗。
失代偿右心衰竭往往合并水钠潴留,表现为中心静脉压升高、肝淤血、腹水和外周水肿。利尿剂可有效改善上述症状。临床中对容量不足,尤其心导管测定右心房压力偏低,超声心动图提示左心室严重受压且血压偏低的患者,应谨慎使用利尿剂。常用利尿剂包括襻利尿剂和醛固酮受体拮抗剂。血管加压素V2受体拮抗剂在此类患者中尚缺乏循证医学证据,需在有经验的中心严密监测下使用。应用利尿剂时应监测肾功能和血生化指标避免出现电解质紊乱和血容量下降引起的肾前性肾功能不全。
当外周血氧饱和度<91%或动脉血氧分压<60 mmHg时建议吸氧,使血氧饱和度>92%。
地高辛可改善PAH患者心输出量,但长期疗效尚不清楚。对合并快速型房性心律失常患者可考虑应用地高辛控制心室率。除左心疾病所致肺高血压外,不建议对其他类型肺高血压患者应用血管紧张素转换酶抑制剂(ACEI)/血管紧张素Ⅱ受体阻滞剂(ARB)、β受体阻滞剂、硝酸酯类药物和伊伐布雷定等药物。特殊情况需应用时应严密监测患者血压、心率和症状,避免PAH靶向药物和上述药物合用产生严重不良反应。
缺铁在PAH患者中较为普遍,其可使PAH患者运动耐量下降,病死率增加,并且这种铁缺乏与贫血无关。铁缺乏患者可考虑铁替代治疗[76],推荐静脉注射铁剂。
推荐意见:女性PAH患者应避免妊娠(Ⅰ,C)。
PAH患者应定期接种流感和肺炎疫苗(Ⅰ,C)。
应对PAH患者进行心理疏导(Ⅰ,C)。
对接受药物治疗的PAH患者建议在专人指导下进行运动康复锻炼(Ⅱa,C)。
WHO心功能Ⅲ~Ⅳ级且动脉血氧分压<60 mmHg的PAH患者乘坐飞机时建议吸氧(Ⅱa,C)。
PAH患者进行择期手术建议尽可能选择硬膜外麻醉(Ⅱa,C)。
PAH患者运动锻炼时以不引起明显不适为宜(Ⅱb,C)。
合并右心功能不全和液体潴留的PAH患者应给予利尿剂治疗(Ⅰ,C)。
IPAH、遗传性PAH及药物相关PAH可考虑长期口服抗凝剂治疗(Ⅱb,C)。
合并矛盾性栓塞的艾森曼格综合征、合并肺动脉原位血栓形成的PAH患者需酌情抗凝治疗(Ⅱb,C)。
PAH患者应纠正贫血和/或缺铁状态(Ⅱb,C)。
PAH患者不建议使用ACEI、ARB、β受体阻滞剂及伊伐布雷定,除非合并相关疾病如高血压、冠心病或左心功能衰竭等(Ⅲ,C)。
需强调,只有急性肺血管扩张试验阳性的PAH患者可单独使用大剂量钙通道阻滞剂治疗,心率偏快首选地尔硫,心率偏慢则首选硝苯地平或氨氯地平。治疗此类PAH患者所需靶剂量往往较大:硝苯地平120~240 mg/d,地尔硫240~720 mg/d,氨氯地平20 mg/d[7]。先给予常规起始剂量,观察患者血压、心律、心率、心电图及症状变化,逐渐增加至最大耐受剂量,并定期随访。至少每3个月1次超声心动图检查。建议服药1年后复查右心导管,如患者WHO心功能稳定在Ⅰ、Ⅱ级,右心结构和功能基本正常,右心导管测定肺动脉压力正常或接近正常(mPAP≤30 mmHg),可判断患者对钙通道阻滞剂治疗持续敏感,可继续长期治疗。如不满足上述标准,需考虑逐渐转换为PAH靶向药物治疗。
推荐意见:IPAH、遗传性PAH及药物相关PAH患者如急性肺血管扩张试验阳性,应给予大剂量钙通道阻滞剂治疗(Ⅰ,C)。
建议对接受大剂量钙通道阻滞剂治疗的IPAH、遗传性PAH及药物相关PAH患者密切随访,治疗3~4个月后全面再评价(包括右心导管检查)(Ⅰ,C)。
对WHO心功能Ⅰ、Ⅱ级且血流动力学明显改善甚至接近正常的IPAH、遗传性PAH及药物相关PAH患者,建议继续给予大剂量钙通道阻滞剂治疗(Ⅰ,C)。
对WHO心功能Ⅲ、Ⅳ级或血流动力学改善不明显的PAH患者,建议启动PAH靶向药物治疗(Ⅰ,C)。
对未接受急性肺血管扩张试验或结果阴性的患者,不建议应用大剂量钙通道阻滞剂治疗(Ⅲ,C)。
内皮素系统异常激活是PAH发生发展的重要机制之一[77]。内皮素-1主要通过与肺血管壁上的内皮素受体A和B结合发挥肺血管收缩和促平滑肌细胞有丝分裂的作用。内皮素受体拮抗剂通过阻断内皮素-内皮素受体信号传导发挥治疗PAH的作用。需注意,由于内皮素受体拮抗剂有潜在致畸作用,服用此类药物需严格避孕。
波生坦:波生坦是一种双重内皮素受体拮抗剂,可同时拮抗内皮素A和B受体。BREATHE研究证实波生坦可改善IPAH、结缔组织病相关PAH和艾森曼格综合征患者的运动耐量、心功能、血流动力学参数并延缓到达临床恶化时间[78]。在我国波生坦治疗PAH疗效和安全性也得到Ⅳ期临床试验和其他一系列临床试验支持[79,80,81]。此外,在欧美波生坦还有治疗儿童PAH的适应证。波生坦引起肝脏转氨酶升高的发生率为6%~10%,且有导致贫血和外周浮肿的风险。治疗期间应监测肝功能和血常规,尤其是治疗开始时的前3~6个月。
安立生坦:安立生坦是一种高选择性内皮素A受体拮抗剂,多项随机对照试验证实安立生坦单药治疗可显著改善PAH患者的症状、运动耐量、血流动力学指标并延缓到达临床恶化时间[82]。此外,AMBITION研究显示安立生坦与他达拉非起始联合治疗临床失败事件发生风险仅为两个单药治疗组合并后的50%,且显著降低PAH导致的再次住院率[12]。安立生坦治疗国人PAH的研究证实,其可改善患者的运动耐量和心功能分级,降低肺动脉收缩压和NT-proBNP水平[83]。安立生坦最常见的不良反应是外周水肿,大多数患者为轻到中度,仅有1.6%的患者长期服用安立生坦会发生重度外周水肿。服用安立生坦无需常规监测肝功能。
马昔腾坦:马昔腾坦是一种新型组织靶向性并具有高度亲脂性的双重内皮素受体拮抗剂。SERAPHIN研究结果显示马昔腾坦可显著延缓PAH患者到达临床恶化进程,并能改善患者心功能分级、运动耐量和血流动力学参数[84]。与单药治疗相比,序贯联合马昔腾坦治疗可显著降低PAH患者恶化/死亡风险[85]。马昔腾坦严重不良反应为贫血,需严密监测血常规,无需常规监测肝功能。
肺血管包含大量5型磷酸二酯酶,而5型磷酸二酯酶是环磷酸鸟苷(cGMP)的降解酶,其抑制剂可通过NO/cGMP通路发挥血管舒张作用。目前5型磷酸二酯酶抑制剂主要包括西地那非、他达拉非和伐地那非。
西地那非:西地那非是首个批准用于PAH治疗的5型磷酸二酯酶抑制剂,多项随机对照试验证实了其治疗PAH的有效性和安全性[86,87]。一项随机对照试验发现在已静脉泵入依前列醇的患者中联合西地那非可进一步改善患者6 min步行距离和延缓到达临床恶化时间[88]。SUPER-2研究结果显示,西地那非单药治疗3年后60%的患者病情稳定,46%的患者6 min步行距离改善[89]。多项临床研究证实西地那非可改善我国PAH患者症状和心功能,安全性和耐受性均较好[90,91,92]。西地那非常见不良反应主要源于其血管舒张作用(如头痛、潮热和鼻衄)和对其他非5型磷酸二酯酶的抑制作用(肌肉疼痛和视觉障碍)等。上述不良反应往往是轻至中度,且具有剂量依赖性,绝大部分患者可逐渐耐受。
他达拉非:他达拉非是目前上市的5型磷酸二酯酶抑制剂中唯一的长效制剂。多项随机对照临床试验证实其可显著改善PAH患者的运动耐量、症状、血流动力学参数和到达临床恶化时间[93,94]。AMBITION研究也证实了他达拉非和安立生坦两种长效PAH靶向药物初始联合治疗的疗效和安全性[10]。他达拉非的不良反应与西地那非相似。
伐地那非:伐地那非也是一种高选择性5型磷酸二酯酶抑制剂。伐地那非治疗PAH的循证医学证据主要源于EVALUATION研究。该研究是在我国PAH患者中开展的随机双盲、安慰剂对照临床试验,结果显示伐地那非可显著改善PAH患者的运动耐量、心功能分级和血流动力学参数,且耐受性良好[95,96]。伐地那非的不良反应与西地那非类似。
西方国家已批准西地那非和他达拉非用于成人PAH的治疗。尽管我国暂无5型磷酸二酯酶抑制剂治疗PAH的适应证,但由于其疗效可靠、价格便宜,已成为我国PAH的一线治疗药物。使用5型磷酸二酯酶抑制剂过程中应避免与硝酸酯类和鸟苷酸环化酶激动剂等药物合用,以免引起严重低血压。
利奥西呱是一种新型的可溶性鸟苷酸环化酶激动剂,可单独或与NO协同提高血浆中cGMP水平。利奥西呱是目前唯一具备PAH和CTEPH双适应证的靶向药物。PATENT-1研究表明利奥西呱可显著改善PAH患者6 min步行距离、血流动力学参数和心功能分级,降低NT-proBNP水平,并延缓到达临床恶化时间[97]。PATENT-2是PATENT-1的开放期延伸研究,经过1年随访,服用利奥西呱患者的临床获益得以延续[98]。利奥西呱治疗结缔组织病相关PAH[99]和手术纠正后的先天性心脏病相关PAH[100]疗效和安全性均良好,其中结缔组织病相关PAH患者2年生存率可达93%[99]。
利奥西呱的常见不良反应:消化道症状(恶心、呕吐、腹泻)最常见(49%),约9%的患者出现低血压,6%的患者出现咯血。大多数患者不良反应为轻至中度,约11%的患者因无法耐受而停药。利奥西呱禁忌与5型磷酸二酯酶抑制剂联用,既往反复咯血的患者慎用。
前列环素可刺激腺苷酸环化酶,使平滑肌细胞内cAMP浓度升高,进而扩张血管。前列环素是目前最强力的内源性血小板聚集抑制剂,且具有细胞保护和抗增殖作用。研究发现PAH患者肺动脉中前列环素合成酶表达减少且尿中前列环素代谢产物降低,表明PAH患者前列环素代谢通路下调。已有多种人工合成的前列环素用于治疗PAH,这些药物尽管药代动力学特征不同,但药效学作用相似。
依前列醇:依前列醇是首个人工合成的前列环素类似物,半衰期短(3~5 min),需要应用持续输注装置通过深静脉持续泵入。多项随机对照试验证实了依前列醇治疗WHO心功能Ⅲ、Ⅳ级的PAH患者的疗效和安全性[101,102,103]。依前列醇是目前唯一经随机对照试验证实可降低PAH病死率的药物,可将死亡风险降低70%[104]。依前列醇的起始剂量一般为2~4 ng·kg-1·min-1,目标剂量一般为20~40 ng·kg-1·min-1,最高可达100 ng·kg-1·min-1以上,应根据患者耐受性制定个体化治疗方案。依前列醇是目前WHO心功能Ⅳ级PAH患者的首选治疗药物。严重不良反应主要包括输送系统异常、局部感染、导管阻塞和败血症。由于依前列醇半衰期极短,突然停药可能出现病情加重、恶化甚至死亡。目前已有改良的依前列醇剂型,室温下稳定时间明显延长(可达8~12 h)。该药即将在我国上市。
伊洛前列素:伊洛前列素是一种化学性质稳定的前列环素类似物,为可雾化吸入剂型,也可静脉泵入。吸入伊洛前列素起效迅速,肺血管选择性好,对体循环影响较小。随机对照试验显示吸入伊洛前列素可显著改善PAH和CTEPH患者的症状、运动耐量和血流动力学参数。由于吸入伊洛前列素起效快速(2~5 min),不仅可作为急性肺血管扩张试验用药[7,56],也可用于肺动脉高压危象的抢救[105,106]。吸入伊洛前列素需配备合适的雾化吸入装置(推荐压缩雾化器),以便雾化颗粒高效地沉积于肺泡。吸入伊洛前列素常见的不良反应包括面部潮热、下颌疼痛、低血压和咳嗽(气道高反应状态)。伊洛前列素亦可通过静脉泵入,用于治疗严重右心衰竭的PAH或CTEPH。
曲前列尼尔:曲前列尼尔是一种在室温下相对稳定、半衰期较长的人工合成前列环素。曲前列尼尔有多种剂型,可通过皮下或静脉持续注射,也可通过吸入或口服给药。临床研究证实皮下注射或雾化吸入曲前列尼尔均能显著改善PAH患者的运动耐量、血流动力学参数和症状[107]。皮下及静脉注射起始剂量一般为1.25 ng·kg-1·min-1,根据患者耐受程度逐渐加量,目标剂量一般为20~80 ng·kg-1·min-1。研究显示,能耐受13.8 ng·kg-1·min-1以上的PAH患者其运动耐量优于耐受剂量较低的患者。皮下注射曲前列尼尔最常见的不良反应为注射部位疼痛和消化系统症状,其次为面部潮热和头痛等。其中注射部位疼痛和消化道症状是我国患者停药的最主要原因。对出现明显不良反应的患者可考虑减缓加量速度,并适当对症治疗。研究证实口服曲前列尼尔可改善既往无背景治疗PAH患者的运动耐量[108]。
贝前列素:贝前列素是首个化学性质稳定的口服前列环素类似物。在欧美进行的随机对照试验显示贝前列素治疗3~6个月可以改善IPAH患者的6 min步行距离[109,110,111],但其长期疗效尚未确认。该药目前已在韩国和日本获得治疗PAH适应证。
司来帕格:司来帕格是一种口服选择性前列环素IP受体激动剂,尽管其与其代谢产物具有和内源性前列环素相似的作用模式,但其与前列环素的药理学机制不同。GRIPHON研究纳入1 156例初治或既往有背景治疗的PAH患者,结果显示司帕来格可显著降低PAH患者的综合临床事件终点(致残率及致死率)[112]。亚组分析显示,与5型磷酸二酯酶抑制剂单药相比,司来帕格两药序贯联合治疗可使PAH患者恶化/死亡风险降低42%;与内皮素受体拮抗剂单药相比,司来帕格两药序贯联合治疗可使恶化/死亡风险降低34%;与5型磷酸二酯酶抑制剂联合内皮素受体拮抗剂相比,司来帕格三药序贯联合治疗可使PAH患者恶化/死亡风险降低37%[113]。司帕来格的不良反应和其他前列环素类药物相似,主要为头痛和消化系统症状。
尽管近年来PAH药物治疗取得巨大进展,但患者长期预后仍不理想。对于PAH这种明确有多个致病通路的疾病,理论上联合治疗较单药治疗效果更好。PAH靶向药物联合应用有序贯联合治疗和起始联合治疗两种策略。近年发布的多项随机对照试验结果显示,序贯联合治疗和起始联合治疗均可显著减少PAH患者临床恶化事件发生[10,85,88,108,113]。因此,除PAH危险分层为低危的患者、老年患者和疑诊PVOD/PCH患者,危险分层为中危或高危的患者均推荐联合治疗。随着PAH靶向药物的增多以及药费的显著降低,我国PAH患者接受联合治疗的比例显著提升,但能起始接受联合治疗或接受充分强度联合治疗患者的比例仍非常低。
肺动脉高压靶向药物的类型、推荐用法和不良反应见表6,治疗流程见图2。
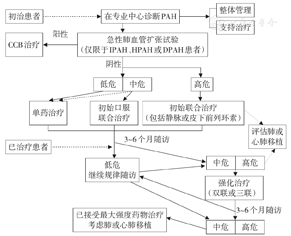
PAH:肺动脉高压,CCB:钙通道阻滞剂,IPAH:特发性肺动脉高压,HPAH:遗传性肺动脉高压,DPAH:药物相关肺动脉高压;实线为明确推荐,虚线为可选推荐

PAH靶向药物的类型、推荐用法和不良反应
PAH靶向药物的类型、推荐用法和不良反应
| 药物 | 适应证 | 推荐用法 | 常见不良反应 | |
|---|---|---|---|---|
| 内皮素受体拮抗剂 | ||||
| 波生坦 | PAH | 口服:成人62.5~125 mg,每日2次;儿童2 mg·kg-1·d-1,分2次口服 | 转氨酶增高 | |
| 安立生坦 | PAH | 口服:成人5~10 mg,每日1次;儿童1.25 ~2.5 mg,每日1次 | 头痛,外周浮肿 | |
| 马昔腾坦 | PAH | 口服:成人10 mg,每日1次;儿童暂无推荐 | 贫血,外周浮肿 | |
| 5型磷酸二酯酶抑制剂 | ||||
| 西地那非 | 暂无 | 口服:成人20~80 mg,每日3次;儿童,年龄-1·d-1,分3次口服;体重20 kg, 20 mg,每日3次 | 潮热、视觉障碍 | |
| 他达拉非 | 暂无 | 口服:成人40 mg,每日1次,推荐10~20 mg,每日1次起始;儿童2.5~10 mg,每日1次 | 潮热、肌痛 | |
| 伐地那非 | 暂无 | 口服:成人5~10 mg,每日2次;儿童1.25~2.5 mg,每日2次 | 潮热、肌痛 | |
| 鸟苷酸环化酶激动剂 | ||||
| 利奥西呱 | PAH和CTEPH | 口服:成人1 mg,每日3次起始,逐渐加量至2.5 mg,每日3次;儿童禁忌使用 | 消化道症状、咯血 | |
| 人工合成前列环素类似物 | ||||
| 依前列醇 | PAH | 静脉泵入:2~4 ng·kg-1·min-1起始,一般推荐剂量20~40 ng·kg-1·min-1,最大可至100 ng·kg-1·min-1以上 | 头痛、消化道症状、输注路径感染 | |
| 伊洛前列素 | PAH | 雾化吸入:成人10~20 μg,每6 h 1次;儿童暂无推荐静脉泵入:0.5~4.0 ng·kg-1·min-1 | 头痛、低血压、咳嗽 | |
| 曲前列尼尔 | PAH | 皮下和静脉:1.25 ng·kg-1·min-1起始,逐渐增加至推荐剂量20~40 ng·kg-1·min-1 | 输注部位疼痛、头痛和消化道症状 | |
| 贝前列素 | 暂无 | 口服:成人40~120 μg,每日4次;儿童暂无推荐 | 头痛、消化道症状 | |
| 前列环素IP受体激动剂 | ||||
| 司来帕格 | PAH | 口服:成人200 μg,每日2次,每周上调200 μg至耐受剂量,最大剂量1 600 μg,每日2次;儿童暂无推荐 | 头痛、消化道症状 | |
注:PAH为肺动脉高压,CTEPH为慢性血栓栓塞性肺高血压
推荐意见:根据PAH患者WHO心功能分级推荐的单药治疗、起始联合治疗方案分别见表7、表8,对WHO心功能Ⅳ级的患者,建议先给予以静脉或皮下前列环素为基础的联合治疗至少3个月,仍然疗效不佳时可考虑肺移植或心肺联合移植。序贯联合治疗方案应根据患者具体情况选择。
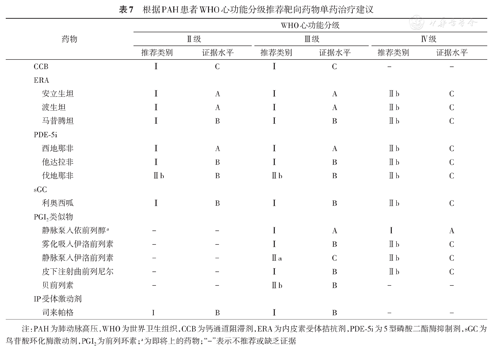
根据PAH患者WHO心功能分级推荐靶向药物单药治疗建议
根据PAH患者WHO心功能分级推荐靶向药物单药治疗建议
| 药物 | WHO心功能分级 | ||||||
|---|---|---|---|---|---|---|---|
| Ⅱ级 | Ⅲ级 | Ⅳ级 | |||||
| 推荐类别 | 证据水平 | 推荐类别 | 证据水平 | 推荐类别 | 证据水平 | ||
| CCB | Ⅰ | C | Ⅰ | C | - | - | |
| ERA | |||||||
| 安立生坦 | Ⅰ | A | Ⅰ | A | Ⅱb | C | |
| 波生坦 | Ⅰ | A | Ⅰ | A | Ⅱb | C | |
| 马昔腾坦 | Ⅰ | B | Ⅰ | B | Ⅱb | C | |
| PDE-5i | |||||||
| 西地那非 | Ⅰ | A | Ⅰ | A | Ⅱb | C | |
| 他达拉非 | Ⅰ | B | Ⅰ | B | Ⅱb | C | |
| 伐地那非 | Ⅱb | B | Ⅱb | B | Ⅱb | C | |
| sGC | |||||||
| 利奥西呱 | Ⅰ | B | Ⅰ | B | Ⅱb | C | |
| PGI2类似物 | |||||||
| 静脉泵入依前列醇a | - | - | Ⅰ | A | Ⅰ | A | |
| 雾化吸入伊洛前列素 | - | - | Ⅰ | B | Ⅱb | C | |
| 静脉泵入伊洛前列素 | - | - | Ⅱa | C | Ⅱb | C | |
| 皮下注射曲前列尼尔 | - | - | Ⅰ | B | Ⅱb | C | |
| 贝前列素 | - | - | Ⅱb | B | - | - | |
| IP受体激动剂 | |||||||
| 司来帕格 | I | B | Ⅰ | B | - | - | |
注:PAH为肺动脉高压,WHO为世界卫生组织,CCB为钙通道阻滞剂,ERA为内皮素受体拮抗剂,PDE-5i为5型磷酸二酯酶抑制剂,sGC为鸟苷酸环化酶激动剂,PGI2为前列环素;a为即将上的药物;"-"表示不推荐或缺乏证据

根据PAH患者WHO心功能分级推荐靶向药物起始联合治疗方案
根据PAH患者WHO心功能分级推荐靶向药物起始联合治疗方案
| 起始联合治疗方案 | WHO心功能分级 | |||||
|---|---|---|---|---|---|---|
| Ⅱ级 | Ⅲ级 | Ⅳ级 | ||||
| 推荐类别 | 证据水平 | 推荐类别 | 证据水平 | 推荐类别 | 证据水平 | |
| 安立生坦+他达拉非 | Ⅰ | B | Ⅰ | B | Ⅱb | C |
注:PAH为肺动脉高压,WHO为世界卫生组织
肺高血压患者由于各种诱因出现右心衰竭进展或接受外科手术时需转入重症监护室治疗。除监测常规生命体征外,还应监测中心静脉压、中心静脉血氧饱和度和血乳酸水平。若患者中心静脉氧饱和度<60%、血乳酸水平上升和尿量减少,预示右心衰竭恶化。部分患者需进行床旁漂浮导管监测,以便对血流动力学进行全面评估。肺高血压合并重症右心衰竭的治疗原则包括治疗诱发因素(如贫血、心律失常、感染或其他合并症)、优化容量管理(通常应用静脉利尿剂)、降低右心室后负荷(对于PAH和CTEPH患者,首选静脉或皮下或吸入前列环素类似物,可联合其他PAH靶向药物治疗)、应用正性肌力药物(首选多巴酚丁胺,对于心率偏快的患者可选择左西孟旦)改善心输出量以及维持体循环血压(首选去甲肾上腺素和多巴胺)等。气管插管可导致血流动力学不稳定,右心衰竭患者应尽量避免。
肺高血压合并严重右心衰竭且药物治疗效果不佳时可考虑使用体外膜肺氧合(ECMO)进行救治,但需提前明确下一步治疗方向,过渡到恢复,或过渡到肺移植或心肺联合移植。建议ECMO仅用于明确有恢复机会或等待移植的患者。
球囊扩张房间隔造口术通过右向左分流降低右心房压力,增加左心室前负荷和心输出量。尽管球囊扩张房间隔造口术会因右向左分流增加导致动脉氧饱和度降低,但心输出量增加可改善体循环氧气运输,并降低交感神经过度兴奋。在有经验的中心,球囊扩张房间隔造口术可作为重症肺动脉高压姑息性治疗手段或肺移植前的过渡性治疗措施[114,115]。该治疗有一定风险,需谨慎选择临床适应证。禁忌证:右心房压力>20 mmHg,静息状态动脉血氧饱和度<85%等。球囊扩张房间隔造口术多采用球囊逐级扩张法,但瘘口再闭塞率高,因而血流动力学改善难以长期维持。新的造口方法包括使用射频消融导管进行房间隔造口或植入带孔封堵器等,他们的疗效和安全性尚有待证实[116]。
经充分的内科药物治疗(至少使用过包括静脉或皮下前列环素类药物在内的联合治疗),仍合并严重血流动力学受损(心指数<2 L·min-1·m-2)、运动耐量显著降低(6 min步行距离<350 m)和明显右心衰竭征象的肺高血压患者可考虑行肺移植或心肺联合移植[117,118]。对于终末期PAH和慢性呼吸系统疾病所致肺高血压患者,一般选择肺移植即可。对于复杂先天性心脏病和左心疾病所致肺高血压则需考虑心肺联合移植或单纯心脏移植治疗。PVOD和PCH由于缺乏有效治疗药物,多数患者病情进展迅速,确诊后应及早进行肺移植评估。目前国内外针对肺高血压患者一般选择双肺移植治疗。IPAH患者肺移植术后3个月的病死率(23%)显著高于因慢性阻塞性肺疾病或囊性纤维化(均为9%)行肺移植治疗的患者,主要原因是约1/3的PAH患者肺移植术后左心室充盈压突然升高,诱发左心衰竭[119]。最近有中心报道,在ECMO支持下,PAH患者肺移植术后3个月、1年和5年生存率分别为93%、90%和87%[120]。我国终末期PAH接受肺移植治疗的数量较少,有研究显示18例IPAH患者行双肺移植术后1年和3年生存率分别为77.8%和72.2%[121]。
我国有学者开展了一系列经皮肺动脉去神经术治疗对药物治疗反应不佳的肺动脉高压患者的临床试验,发现部分患者心功能和血流动力学参数有所改善[122,123]。不过该技术具体应用范围和疗效仍有待进一步证实。
对重症PAH患者重症监护和房间隔造口术及肺移植治疗的建议详见表9。
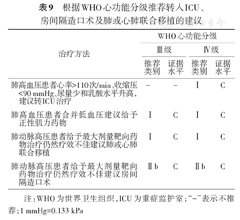
根据WHO心功能分级推荐转入ICU、房间隔造口术及肺或心肺联合移植的建议
根据WHO心功能分级推荐转入ICU、房间隔造口术及肺或心肺联合移植的建议
| 治疗方法 | WHO心功能分级 | |||
|---|---|---|---|---|
| Ⅲ级 | Ⅳ级 | |||
| 推荐类别 | 证据水平 | 推荐类别 | 证据水平 | |
| 肺高血压患者心率>110次/min、收缩压 | - | - | Ⅰ | C |
| 肺高血压患者合并低血压建议给予正性肌力药物 | Ⅰ | C | Ⅰ | C |
| 肺动脉高压患者给予最大剂量靶向药物治疗仍然疗效不佳建议肺或心肺联合移植 | Ⅰ | C | Ⅰ | C |
| 肺动脉高压患者给予最大剂量靶向药物治疗仍然疗效不佳建议房间隔造口术 | Ⅱb | C | Ⅱb | C |
注:WHO为世界卫生组织,ICU为重症监护室;"-"表示不推荐;1 mmHg=0.133 kPa
心律失常是肺高血压的常见并发症,尤其当右心结构重构明显或合并电解质紊乱时容易发生。与左心疾病相比,恶性室性心律失常如室性心动过速、心室扑动和心室颤动在肺高血压患者中非常少见。在132例发生心脏骤停的PAH患者中,仅8%发生心室颤动[124]。另一项研究对231例PAH和CTEPH患者随访6年,未观察到恶性室性心律失常的发生情况,但室上性心律失常的年发生率仅为2.8%[125]。我国开展的一项IPAH心律失常的观察性研究显示,室上性心动过速6年累积发生率为15.8%[38]。持续性房性心律失常尤其是房颤和房扑提示预后不佳。室上性心律失常一旦发生应积极复律治疗,药物难以复律时可考虑电复律或射频消融。
咯血是肺高血压患者常见并发症,也是导致患者病情加重甚至死亡的重要诱因。咯血可来源于肺动脉畸形或代偿扩张的支气管动脉,一些特殊类型肺高血压如遗传性出血性毛细血管扩张症合并PAH、艾森曼格综合征和CTEPH更易咯血。咯血严重程度差异较大,大部分患者为少至中量咯血,一般可自行终止,无需特殊处理。部分患者可发生严重的大咯血或迁延多日的咯血,可导致窒息、失血性休克、肺不张、严重肺部感染以及猝死等危及生命的情况。有研究报道肺高血压患者咯血发生率为1%~6%[7]。对于肺动静脉瘘破裂导致的严重出血需行肺动静脉瘘介入封堵治疗,对于支气管动脉迂曲扩张破裂所致的严重咯血可考虑支气管动脉栓塞治疗。无论肺动静脉瘘介入封堵还是支气管动脉栓塞治疗均有导致肺动脉高压病情加重的风险,一旦发生应积极治疗。
肺高血压的机械并发症通常与肺动脉进行性扩张有关,包括肺动脉瘤样扩张导致破裂和夹层,压迫胸腔组织如左冠状动脉主干、肺静脉、主支气管和喉返神经等。其症状和体征与具体压迫的部位相关,包括胸痛(类似心绞痛)、单侧声带麻痹导致的声音嘶哑、局部肺水肿和猝死等。增强胸部CT是诊断肺动脉扩张及其他血管组织受累最重要的方法。肺高血压合并肺动脉瘤样扩张、假性动脉瘤和夹层尚缺乏有效的治疗方法。对于冠状动脉左主干显著受压并表现为明显心绞痛的患者可考虑经皮冠状动脉介入治疗。
先天性心脏病相关PAH是PAH的一个重要亚类,目前缺乏可靠的流行病学数据。欧洲一项注册登记研究提示,成年先天性心脏病患者中PAH患病率为5%~10% [126]。我国由于医疗水平所限,相当多的先天性心脏病未能及时诊断和治疗,部分患者甚至进展为艾森曼格综合征,因而先天性心脏病相关PAH已成为我国PAH患者中最常见的疾病亚类。
根据先天性心脏病相关PAH的解剖和病理生理学特点进行临床分类,可分为4类:(1)艾森曼格综合征:包括所有初始为体循环到肺循环心内或心外分流,后因肺血管阻力升高而逆转为肺循环到体循环分流或双向分流的先天性心脏病,常合并紫绀、继发性红细胞增多症和多器官受累。(2)体肺分流性先天性心脏病:分为可矫治和不可矫治两个亚类,可矫治指中到大型缺损、肺血管阻力轻到中度升高、以体循环向肺循环分流为主,是否合并紫绀不是主要特征;不可矫治指目前无介入封堵或外科手术修补的适应证。(3)PAH并发先天性心脏病:小缺损合并肺血管阻力显著升高,且单用缺损无法解其升高;临床特征与IPAH相似。禁忌关闭此类缺损。(4)先天性心脏病术后PAH:缺损修补或介入封堵后PAH仍持续存在,数月或数年内再发PAH。此型病情易进行性加重。
先天性心脏病相关PAH的治疗应根据缺损性质、大小和血流动力学特点判断。对有矫治适应证的患者应及早进行缺损的修补或介入封堵治疗,避免长期大量分流导致不可逆的肺血管重构。
手术可行性判断:左向右分流型先天性心脏病相关PAH手术时间窗相对较宽,需根据缺损的大小和性质综合判断。决定患者术后结局的两个关键因素为手术年龄和术前肺血管阻力。肺血管阻力和肺血管阻力/体循环阻力比值是临床常用的判断可行性指标,这两个指标越高,术后残余PAH的风险越高[7,127,128]。但目前尚缺乏国际统一判断标准。
基础治疗:对存在明显紫绀的艾森曼格综合征患者推荐家庭氧疗,尽管目前尚无充分循证医学证据支持吸氧可改善艾森曼格综合征患者的生存率或降低血红蛋白水平[129]。抗凝治疗尚存争议。应尽量避免选择含雌激素的口服避孕药,因雌激素可增加血栓栓塞的风险。
PAH治疗:不能修补的先天性心脏病相关PAH或术后PAH患者推荐使用PAH靶向药物治疗。治疗目标主要为缓解症状和改善预后。部分处于临界手术指征的先天性心脏病相关PAH患者经PAH靶向药物治疗后甚至可获得手术治疗机会[130]。
中国系统性红斑狼疮协作组(CSTAR)注册研究表明,系统性红斑狼疮相关PAH的患病率约为3.8%,系统性硬化症相关PAH患病率约为11%,另外原发性干燥综合征、混合性结缔组织病、皮肌炎、类风湿关节炎亦可导致PAH[132]。我国近30年系统性红斑狼疮死因分析发现,PAH是继神经精神性狼疮、狼疮性肾炎之后第3位常见死亡原因[133,134,135,136]。国外近30年系统性硬化症死因分析发现,不伴肺间质纤维化的PAH致死率亦明显升高(20%升至30%)[137,138]。北京协和医院的单中心研究显示,系统性红斑狼疮、原发性干燥综合征、系统性硬化症合并PAH患者的5年生存率分别为61.0%、43.9%、64.8%,远低于不合并PAH的结缔组织病患者[139]。早期诊断、早期干预才有望改善我国结缔组织病相关PAH患者的预后。
首先指风湿科医师定期对结缔组织病患者进行PAH相关筛查,在尚无明显症状的亚临床期诊断出潜在的PAH,其次指心内科或呼吸科医师对已诊断的PAH患者常规进行结缔组织病相关指标的筛查,并在风湿科医师参与下确诊合并的结缔组织病。早期诊断的意义在于:(1)及时给予PAH靶向治疗;(2)尽早启动结缔组织病的免疫抑制治疗,对阻止乃至逆转PAH进展十分重要。
国际上建议结缔组织病患者一旦出现活动后气短、心悸、胸痛、咯血等症状应尽快进行PAH筛查。如不能诊断PAH但症状持续或加重,应在3个月内重复进行检查。而对无症状的系统性硬化症患者建议每年进行1次PAH筛查[129]。我国也建议对系统性硬化症进行PAH筛查。CSTAR研究显示,在我国系统性红斑狼疮也是PAH的高危因素,对病情活动(尤其存在心包炎、胸膜炎时)、抗核糖核蛋白(RNP)抗体阳性和/或存在雷诺现象的系统性红斑狼疮患者应重视PAH的早期筛查[135]。我国相关研究还发现原发性干燥综合征是PAH的高危因素,对合并雷诺现象、肝脏损伤、心包炎、高滴度类风湿因子的原发性干燥综合征患者也应重视PAH的早期筛查。
心内科或呼吸科医师一旦诊断PAH患者,应尽早进行结缔组织病初筛:(1)问诊是否有雷诺现象、手指肿胀、皮疹、关节肿痛、口腔溃疡、口干、眼干等;(2)常规检查是否有血细胞减少、尿红细胞或尿蛋白阳性、高球蛋白血症、低补体血症等;(3)自身抗体检查包括抗核抗体、抗可提取的核抗原抗体、抗磷脂抗体等。如有提示结缔组织病的临床表现或实验室检查异常,应尽快请风湿科医师会诊以明确诊断,指导治疗。
PAH的筛查流程可参照图1。
结缔组织病相关PAH确诊后不仅应评估PAH的严重程度,也应对结缔组织病活动性进行评估,并在治疗过程中密切随访,根据随访结果指导调整治疗方案。系统性红斑狼疮评估体系分为整体评估(如系统性红斑狼疮活动指数)和器官/系统评估(如不列颠群岛狼疮评估小组量表)。原发性干燥综合征的评估体系为欧洲抗风湿病联盟干燥综合征疾病活动指数(ESSDAI)。而系统性硬化症的评估体系则是对皮肤硬化、肺间质病变等纤维化表型的评估。但以上体系均未对PAH进行评估。对结缔组织病相关PAH的危险分层评估参照本指南中PAH风险评估和随访部分。
结缔组织病相关PAH确诊及全面评估后,应根据患者具体情况制定治疗目标及方案。结缔组织病相关PAH的治疗目标是改善患者生活质量,最大程度地改善患者预后。短期目标是延缓到达临床恶化时间,推荐双重达标:(1)结缔组织病病情缓解,以医师整体评估疾病活动评分(physician global assessment,PGA)<1分表示结缔组织病处于临床缓解状态;(2)PAH临床达标或处于低危状态。2015年CSTAR发表的"中国成人系统性红斑狼疮相关肺动脉高压诊治共识"率先提出双重达标的理念,就是希望风湿免疫科医师、心内科/呼吸科医师能够同时重视结缔组织病基础疾病及PAH的治疗[139]。
结缔组织病相关PAH基础疾病的治疗十分重要,对改善和稳定PAH患者病情至关重要,但目前循证医学证据非常有限。首先应针对不同结缔组织病制定相应地治疗策略。活动性系统性红斑狼疮且PAH未达标的患者,往往采用大剂量或冲击剂量的诱导缓解方案;以血管病变为主的原发性干燥综合征相关PAH或系统性硬化症相关PAH则多选择中或小剂量糖皮质激素。糖皮质激素联合免疫抑制剂,特别是环磷酰胺(CTX)是应用最多的方案,可改善系统性红斑狼疮相关PAH患者的功能状态、血流动力学及长期预后。霉酚酸酯(MMF)、硫唑嘌呤(AZA)、环孢素(CsA)、他克莫司(FK506)、甲氨蝶呤(MTX)或羟氯喹(HCQ)等方案在结缔组织病相关PAH中均有应用,但需要个体化,由风湿科医师确定相应的诱导缓解、巩固缓解和维持缓解的序贯治疗。
结缔组织病相关PAH针对PAH的治疗亦十分重要,分为一般治疗和肺血管扩张治疗。几乎所有针对PAH靶向药物的临床试验中均纳入了相当比例的结缔组织病相关PAH,且结缔组织病相关PAH亚组分析中亦能获得阳性结果。结缔组织病相关PAH的治疗可参照本指南中PAH靶向药物治疗部分。
门脉高压相关肺高血压是门脉高压合并肺血流量增加或肺血管重构而引起的肺高血压,可伴或不伴肝脏疾病。门脉高压患者1%~5%罹患肺高血压[140],5%~6%肝移植术前评估患者合并肺高血压[141]。门脉高压相关肺高血压国内报道较少,单中心回顾性研究提示门脉高压相关肺高血压约占肺动脉高压的0.03%,肝硬化是导致我国门脉高压最常见的病因[142]。其发病机制包括门脉高压导致肺循环高动力状态、血管活性介质失衡、凝血和纤溶异常等。
门脉高压相关肺高血压的症状与体征与其他类型肺高血压相似,包括活动后呼吸困难、右心衰竭等,同时具有门静脉高压及原发肝脏疾病的临床表现,如脾大、黄疸、肝掌、蜘蛛痣、腹水、食管胃底静脉曲张等。经胸多普勒超声心动图可作为门脉高压相关肺高血压的筛查手段,如估测肺动脉收缩压升高,需行右心导管检查确诊,以指导治疗、评估预后。美国梅奥诊所将门脉高压相关肺高血压依据血流动力学特点分为两类[143]:仅有mPAP和心输出量升高但肺血管阻力<3 Wood单位,即肺循环高动力低阻力状态;mPAP和肺血管阻力均升高,即肺血管高阻力状态,此类患者肝移植术后死亡率较高。
门脉高压相关肺高血压的治疗目前尚缺乏随机对照试验的证据,少量病例报道观察PAH靶向药物的疗效,提示波生坦、安立生坦、西地那非、曲前列素和利奥西呱等有可能改善门脉高压相关肺高血压患者的血流动力学状态和运动耐量。由于波生坦有导致血清转氨酶升高的风险,因此服用波生坦期间应密切监测肝脏功能。Child-Pugh分级B或C级的患者应避免使用波生坦。抗凝治疗会增加门脉高压相关肺高血压的出血风险,应避免使用。尽管β受体阻滞剂可降低门脉压力和预防食管胃底静脉出血,但易导致血流动力学状态恶化及运动耐量降低,因此不推荐使用。肝脏移植不能改善门脉高压相关肺高血压患者的预后,血流动力学状态恶化的患者可考虑肺肝或心肺肝联合移植。
门脉高压相关肺高血压患者的预后较IPAH患者更差。美国REVEAL注册登记研究发现,门脉高压相关肺高血压患者2年、5年生存率分别为67%、40%[144],心力衰竭和并发症是最主要的死亡原因。
目前全球血吸虫病波及人口已达2亿,其中10%可能发展为肝脾型血吸虫病,而有近5%的此类患者可发生PAH[145]。因此血吸虫病已成为PAH潜在的致病因素[7]。血吸虫病相关PAH除具有血吸虫病相关临床表现外,其余症状与PAH相似。未经PAH靶向药物治疗的患者3年生存率为85.9%[146]。一项小样本回顾性研究表明,血吸虫病相关PAH患者可以从PAH靶向药物治疗中获益[147]。我国是血吸虫病感染大国,每年新发血吸虫病感染病例约10万,但缺乏血吸虫病相关PAH的报道[148]。
HIV感染是明确的PAH危险因素之一,HIV感染患者PAH患病率为0.46%,较普通人群发生IPAH的风险高2 500倍。西方国家PAH注册登记研究显示,HIV感染相关PAH占PAH的2%~7%[149]。瑞士HIV感染人群研究表明,HIV感染相关PAH发病率由1995年的0.21%降至2006年的0.03%,可能与高活性抗逆转录病毒治疗有关[150]。HIV感染相关PAH的症状、血流动力学参数和预后均与IPAH相似[151]。PAH是HIV感染患者死亡的独立危险因素。
PVOD/PCH是一类极为罕见的肺动脉高压亚类,其组织病理学特点除有肺小动脉重构外,肺小静脉亦发生广泛狭窄或闭塞病变,肺毛细血管往往合并扩张性改变或增殖样改变。西方国家普通人群中PVOD/PCH的发病率为0.1~0.2/百万人年[152],但由于诊断困难,实际发病率可能被低估。由于肺活检风险较高,临床上已很少用于PVOD/PCH的诊断。目前推荐的临床诊断标准为[153]:(1)具有典型的PAH症状和血流动力学特点。(2)胸部高分辨率CT具备以下特征:小叶中心性磨玻璃影;小叶间隔线增粗;纵隔淋巴结肿大。(3)呼吸功能检查肺泡弥散功能显著下降。(4)PAH靶向药物治疗病情好转不明显甚至加重,胸部高分辨率CT提示肺部渗出性病变较前增多。(5)遗传学检测明确患者携带EIF2AK4基因纯合突变或复合杂合突变,并经父母验证确认隐性遗传模式。疑诊PVOD/PCH患者应在严密观察下谨慎使用PAH靶向药物治疗,一旦肺水肿加重应立即停药。确诊患者如果药物治疗效果不佳,需尽快考虑肺移植。PVOD/PCH预后极差,确诊后1年死亡率高达74%[154]。
新生儿持续性肺高血压病因复杂而不明确,常表现为出生后数小时血氧饱和度不稳定和进行性青紫[155]。当患儿缺氧、呼吸窘迫程度与胸片不一致或出现差异性紫绀时应考虑新生儿持续性肺高血压,建议行超声心动图检查观察动脉导管和卵圆孔水平是否存在右向左分流。新生儿持续性肺高血压不以肺动脉压力为诊断标准,而是以肺血管阻力升高、动脉导管及卵圆孔水平的右向左分流为判断依据。新生儿持续性肺高血压的治疗目标是维持体循环血压、降低肺血管阻力、改善组织供氧、使吸氧和机械通气的损伤最小化[156]。除一般支持治疗及强心治疗外,特异性肺血管扩张剂包括吸入型NO、内皮素受体拮抗剂、5型磷酸二酯酶抑制剂和前列环素类似物也可用于新生儿持续性肺高血压的治疗[156]。必要时可以使用ECMO辅助治疗。
肺高血压可发生在新生儿到成人的任一年龄段。胎儿时期肺动脉压与主动脉压均维持在较高水平,出生后肺动脉压生理性下降,足月儿一般在生后2~3个月降至成人水平。如果足月儿出生3个月后右心导管测定mPAP≥25 mmHg,则可确诊肺高血压[157]。荷兰注册登记研究显示,儿童肺高血压发病率为63.7/百万人年,其中IPAH的发病率为0.7/百万人年,患病率为4.4/百万[158]。TOPP研究是目前唯一一项国际多中心儿童(>3岁)肺高血压注册登记研究,结果显示入选的362例肺高血压患儿中PAH占88%,其中IPAH/遗传性PAH和先天性心脏病相关PAH患者分别占57%和36%[159]。依前列醇问世之前,肺高血压患儿的中位生存时间仅有约10个月,新型靶向药物出现后其预后明显改善,IPAH/家族性PAH的2年和5年生存率分别达到90%和75%[158]。
儿童与成人肺高血压在血管结构与功能、遗传学、病程、右心室反应性、靶向药物治疗反应性等方面均存在差异,且肺高血压患儿相关致病因素更多,因此WHO肺高血压分类标准是否适用于儿童患者尚存争议。2011年国际肺血管病研究学院(PVRI)提出巴拿马分类标准,将儿童肺高血压分为10个亚类,但尚未广泛应用[160]。IPAH、遗传性PAH及先天性心脏病相关PAH是儿童最常见的肺高血压类型。
呼吸困难、乏力、发育迟缓是儿童肺高血压的常见症状。儿童肺高血压患者晕厥更常见,而右心衰竭出现较晚,可能在出现右心衰竭之前即发生猝死。儿童肺高血压的病因众多,分类复杂,疑诊时建议到专业儿科肺高血压中心就诊。在诊断思路上,应首先考虑先天性心脏病相关PAH、左心疾病所致肺高血压和呼吸系统疾病所致肺高血压,其次为遗传代谢性疾病,再次排除结缔组织病和CTEPH,排除所有已知病因后再考虑IPAH[161]。
确诊肺高血压及靶向药物治疗前需行右心导管检查,必要时加行左心导管检查,对所有IPAH和先天性心脏病相关PAH患儿均应行急性肺血管扩张试验,可为药物和手术治疗提供参考。儿童急性肺血管扩张试验阳性的判断标准仍存在争议[162]。除了成人常用的Sitbon标准外,还有REVEAL标准,即主要为肺动脉压和肺血管阻力/体循环阻力下降≥20%,且不伴心输出量下降。根据TOPP研究数据显示,更为严格的Sitbon标准和患儿预后相关性最强[159]。为评估治疗效果及预后可重复心导管检查,检查间隔时间根据症状、疗效等指标综合评估。儿童肺动脉高压患者行心导管检查风险较成人高,年龄小(<1岁)、WHO心功能Ⅳ级为主要危险因素。儿童同样存在睡眠呼吸障碍风险,尤其是合并21三体综合征或有明显日间嗜睡或对PAH靶向药物治疗反应差的患儿应行睡眠监测。建议合适年龄的肺高血压患儿行心肺运动试验和6MWT以评估运动耐量、疗效及预后。
肺高血压患儿应行血尿遗传代谢检查,对于发育迟缓、血同型半胱氨酸水平显著升高的患儿,需重点疑诊甲基丙二酸尿症。其他血液学和影像学检查与成人相似。
儿童肺高血压一般支持治疗和策略与成人相似。PAH靶向药物需按照公斤体重给药,但均缺乏儿童专用剂型,应用大多参照成人PAH的治疗经验和数据。
急性肺血管扩张试验阳性且年龄>1岁的患儿可使用钙通道阻滞剂进行治疗。常用药物包括硝苯地平(2~5 mg·kg-1·d-1)、地尔硫(3~5 mg·kg-1·d-1)、氨氯地平(2.5~10 mg/d)。需密切随访,必要时重复急性肺血管扩张试验以判断是否持续阳性。钙通道阻滞剂治疗后症状和血流动力学参数无明显改善者,尽早更换靶向药物治疗。
依前列醇、曲前列素均可改善IPAH患儿血流动力学参数和生活质量,提高其生存率。先天性心脏病患儿围术期如发生PAH危象,雾化吸入伊洛前列素疗效显著,可替代NO[163]。2011年欧洲药监局批准西地那非应用于1~17岁的PAH患儿,推荐剂量为:年龄<1岁,0.5~1 mg/kg,每日3次;体重<20 kg,10 mg,每日3次;体重>20 kg,20 mg,每日3次。他达拉非和伐地那非在儿童患者中应用的报道较少。2009年欧洲批准波生坦用于≥2岁儿童PAH的治疗,推荐剂量为2 mg/kg,后将年龄放宽至1岁。2017年美国食品药品管理局(FDA)也批准了波生坦用于≥3岁的IPAH及先天性心脏病相关PAH患儿,我国研究也证实波生坦治疗中国儿童先天性心脏病相关PAH安全、有效[164,165]。安立生坦和马昔腾坦对PAH患儿的疗效和安全性尚待确认。对足月或接近足月的新生儿持续性肺高血压,吸入NO能迅速降低肺动脉压、改善肺血流,从而减少对ECMO治疗的需求。吸入NO还对先天性心脏病围术期反应性PAH和PAH危象也有较好的治疗效果。肺高血压患儿介入和外科手术治疗适应证和策略也与成人相似。
肺高血压是左心疾病常见的合并症,包括射血分数保留的心力衰竭(HFpEF)和射血分数降低的心力衰竭(HFrEF)以及心脏瓣膜病等患者。慢性心力衰竭一旦合并肺高血压,往往提示症状和运动耐量受损更严重,预后更差[166]。
超声心动图是筛查左心疾病所致肺高血压的重要手段,但确诊仍需行右心导管检查[167]。此类患者禁忌急性肺血管扩张试验,因其有导致左心房压升高甚至诱发肺水肿的风险。长期应用利尿剂的患者PAWP可能正常,HFpEF患者静息状态下PAWP也可能<15 mmHg[168]。液体负荷或运动负荷试验可能有助于发现潜在的左心疾病所致肺高血压,但具体的操作方法及临床价值仍有待证实。
左心疾病所致肺高血压应以治疗基础疾病为主,包括严格的液体管理,药物(包括利尿剂、硝酸酯类药物、血管紧张素Ⅱ转换酶抑制剂、β受体阻滞剂和正性肌力药物等)或手术(左心室辅助装置、瓣膜手术、再同步化治疗或心脏移植)治疗,上述治疗可能因左心室充盈压下降而使肺动脉压力迅速下降[169]。由于缺乏循证医学证据,目前暂不推荐在左心疾病所致肺高血压患者中使用PAH靶向药物。
呼吸系统疾病和/或缺氧所致肺高血压分两类:通气受限为特点的阻塞性肺疾病,以慢性阻塞性肺疾病为主;容量受限为特点的限制性肺疾病,以间质性肺疾病为主。大部分慢性肺疾病相关肺高血压患者的肺动脉压力为轻中度升高,仅少部分肺动脉压力严重升高,即mPAP≥35 mmHg或mPAP≥25 mmHg合并低心输出量(心指数<2.5 L·min-1·m-2)。这部分严重肺高血压在慢性阻塞性肺疾病患者中仅占1%左右,需与PAH合并慢性肺疾病相鉴别。另外,阻塞性睡眠呼吸暂停综合征患者肺高血压患病率为17%~42%[170],临床医师应高度关注。慢性呼吸系统疾病一旦出现肺高血压则意味预后不良。
超声心动图是呼吸系统疾病无创筛选肺高血压的重要手段,但特异度、敏感度有限[171]。右心导管仍是诊断肺高血压的"金标准"。对于慢性阻塞性肺疾病或间质性肺疾病相关肺高血压且合并长期低氧血症的患者,长期氧疗是可选的治疗方法。目前尚缺乏PAH靶向药物治疗此类肺高血压的疗效和预后数据。严重肺部疾病[特发性肺纤维化,用力肺活量(FVC)<70%预计值;慢性阻塞性肺疾病,第1秒用力呼气量(FEV1)<60%预计值]和肺动脉压轻中度升高(25 mmHg<mPAP<35 mmHg)者,不建议进行PAH靶向药物治疗[172],因为在扩张肺血管同时会影响肺气体交换加重缺氧。但对存在与原发肺部疾病不匹配的严重肺高血压患者建议到肺血管疾病区域医疗中心进行个体化评估。严重睡眠呼吸障碍患者,行无创通气长期治疗可改善血流动力学参数和右心功能。
慢性高原缺氧也是导致肺高血压的常见原因,而我国是慢性高原病医疗负担最重的国家[173]。高原性肺高血压主要是由于高原地区空气中含氧量减少,肺泡气氧分压下降,诱发肺小动脉收缩及结构重构、血液稠度增加、血容量和肺血流量增加等机制引起。高原性肺高血压的准确发病率尚不清楚,男性发生风险高于女性,可能与雌激素的保护作用有关。与永久居住在青藏高原的藏族居民相比,移居居民发生肺高血压的风险更高;儿童发生肺高血压的风险要高于成人;吸烟、睡眠呼吸障碍或室内空气污染等均可使高原性肺高血压发生增加[174]。
高原性肺高血压的首要治疗方法是移居至平原地区,大多数患者在离开高原地区后肺高血压可完全或部分缓解。高原性肺高血压的药物治疗分为支持疗法与PAH靶向药物治疗[175,176,177]。支持疗法包括抗凝药,由于肺血管内微血栓形成参与肺高血压的发生,因此对没有抗凝禁忌的患者建议应用抗凝药治疗。靶向药物包括前列环素类似物、内皮素受体拮抗剂及5型磷酸二酯酶抑制剂。有研究证实,波生坦可改善高原性肺高血压患者的肺血流动力学状态及减轻肺水肿。多项研究发现西地那非、伐地那非等5型磷酸二酯酶抑制剂及Rho激酶激动剂法舒地尔也可显著改善高原性肺高血压的症状和血流动力学参数。此外,通心络、红景天等药物可能也有一定疗效,但仍有待于进一步研究证实。
目前认为,CTEPH是由于未溶解的血栓发生机化导致肺血管床阻塞所致。这种纤维机化血栓可造成不同级别肺动脉分支血管的完全阻塞或不同程度的狭窄,并在血管腔内形成条索和分隔[178]。CTEPH不仅存在肺血管腔的机械性狭窄和梗阻,在非机化血栓梗阻区还存在与PAH类似的肺小动脉病变,这也解释了部分患者栓塞面积和肺血管阻力升高不匹配的原因。CTEPH发生发展与以下因素有关[179]:(1)急性肺栓塞病史:尤其是复发性肺栓塞,大面积肺栓塞和特发性肺栓塞;(2)血液学因素:遗传性易栓症、Ⅷ因子、Von Willebrand因子水平升高、非O血型等;(3)其他合并疾病:原发或继发性抗磷脂综合征、脾切除、脑室心房分流手术、起搏器感染、慢性炎性疾病(骨髓炎、炎症性肠病)、甲状腺功能低下及恶性肿瘤等。
CTEPH的诊断标准[179,180]:充分抗凝治疗至少3个月;CT肺动脉造影或肺通气灌注显像或直接肺动脉造影提示存在肺栓塞征象;右心导管测定肺循环血流动力学参数符合PAH诊断标准,以上3个标准须同时满足。CTEPH的治疗方法主要包括肺动脉内膜剥脱术、药物治疗和球囊肺动脉成形术。根据CTEPH患者肺动脉受累情况分级(表10),Ⅰ、Ⅱ级的患者首选肺动脉内膜剥脱术,而Ⅲ、Ⅳ级的患者则首选肺动脉球囊成形术,但也应根据具体临床情况选择合适的治疗策略。

慢性血栓栓塞性肺高血压患者肺动脉受累情况分级
慢性血栓栓塞性肺高血压患者肺动脉受累情况分级
| 分级 | 血栓栓塞位置 |
|---|---|
| 0级 | 无明显机化血栓征象 |
| Ⅰ级 | 机化血栓自主肺动脉开始或一侧肺动脉完全闭塞 |
| Ⅱ级 | 机化血栓自叶一级肺动脉开始 |
| Ⅲ级 | 机化血栓仅起自段一级肺动脉 |
| Ⅳ级 | 机化血栓仅起自亚段一级肺动脉 |
药物治疗:CTEPH患者的基础治疗与PAH相似。对无抗凝禁忌证的患者建议长期充分抗凝治疗,以预防复发性静脉血栓栓塞事件和肺动脉原位血栓形成。PAH靶向药物治疗主要的适应证:无法手术治疗的患者;为适当改善血流动力学状态而行术前准备治疗;肺动脉内膜剥脱术后症状性残余/复发的肺高血压。目前,鸟苷酸环化酶激动剂利奥西呱是唯一具有CTEPH治疗适应证的药物[8]。MERIT研究已证实了马昔腾坦在无法行肺动脉内膜剥脱术或术后残余肺高血压的CTEPH患者中的疗效和安全性,其他PAH靶向药物尽管已广泛用于CTEPH的治疗,但疗效有限[181]。
肺动脉内膜剥脱术:部分CTEPH患者可通过肺动脉内膜剥脱术剥离阻塞在肺动脉内的机化血栓和增生内膜,从而显著改善症状和血流动力学状态,甚至完全治愈。因此,推荐对所有确诊CTEPH患者首先进行肺动脉内膜剥脱术可能性评估。国际多中心CTEPH注册登记研究显示,有56.8%的患者进行了肺动脉内膜剥脱术治疗[182]。影响手术开展及效果的主要因素:(1)肺动脉阻塞部位:越靠近近端血管越容易剥离,目前手术可以剥离到段一级肺动脉水平;(2)肺循环血流动力学状态:术前评价需考虑肺动脉阻塞面积和肺循环血流动力学参数是否匹配,肺血管阻力显著升高患者围术期死亡率也相应增加;(3)合并症情况:有经验的中心肺动脉内膜剥脱围术期死亡率可低至2.2%~3.5%。长期随访结果显示,术后患者的长期生存率明显改善,5年生存率为82%,10年生存率可达75%[183]。建议有条件开展肺动脉内膜剥脱术的中心积极开展此项工作。
改良经皮肺动脉球囊成形术:对不适合行肺动脉内膜剥脱术的CTEPH患者(Ⅲ、Ⅳ级病变为主、合并手术禁忌证、拒绝手术或术后残余肺高血压)可尝试行改良经皮肺动脉球囊扩张治疗[184,185,186]。多项单中心开放性研究显示,逐步、多次经皮肺动脉球囊扩张治疗不但能显著改善CTEPH患者的血流动力学参数和症状,还能有效减少围术期并发症。长期随访结果显示,改良经皮肺动脉球囊扩张治疗后5年生存率可达95%以上。术中最常见并发症为肺血管机械损伤所致的咯血或夹层,术后常见并发症为再灌注性肺水肿和对比剂肾病等。
未知的多种发病机制所致肺高血压病因复杂。可能的机制包括:肺血管收缩、增生性血管病变、外源性压迫、内源性闭塞、高心输出量、血管闭塞和左心衰竭等。血液系统疾病如慢性溶血性贫血、慢性骨髓增生性疾病既可直接导致肺高血压,又可因其治疗方法(如脾切除、长期输血引起铁过剩、使用达沙替尼药物等)引起或加重肺高血压。因此第五大类肺高血压目前以明确诊断为主,治疗主要针对原发疾病,建议转到相应专科进行系统诊治。目前PAH靶向药物尚未批准用于此类疾病的治疗。
专家组成员(按姓氏笔画排序):王岚(同济大学附属上海市肺科医院),王勇(首都医科大学附属北京世纪坛医院),王晓建(中国医学科学院阜外医院),毛毅敏(河南科技大学第一附属医院),田庄(北京协和医院),朱鲜阳(北部战区总医院),伍贵富(中山大学附属第八医院),刘岽(上海交通大学医学院附属瑞金医院),刘倩倩(中国医学科学院阜外医院),刘盛(中国医学科学院阜外医院),纪求尚(山东大学齐鲁医院),李江(中南大学湘雅二医院),李梦涛(北京协和医院),李静惠(中国医学科学院阜外医院),吴炳祥(哈尔滨医科大学附属第二医院),余再新(中南大学湘雅医院),沈节艳(上海交通大学医学院附属仁济医院),张刚成(武汉亚洲心脏病医院),张抒扬(北京协和医院),张卓莉(北京大学第一医院),张晓(广东省人民医院),张海峰(南京医科大学第一附属医院),张曹进(广东省人民医院),张尉华(吉林大学白求恩第一医院),张端珍(北部战区总医院),范粉灵(西安交通大学第一附属医院),周达新(复旦大学附属中山医院),周玮(宁夏医科大学总医院),郑泽琪(南昌大学第一附属医院),赵勤华(同济大学附属上海市肺科医院),荆志成(中国医学科学院阜外医院),胡清华(华中科技大学同济医学院附属同济医院),姜睿(中国医学科学院阜外医院),姚桦(广东省人民医院),顾虹(首都医科大学附属北京安贞医院),黄岚(陆军军医大学新桥医院),黄玮(重庆医科大学附属第一医院),曹云山(甘肃省人民医院),程江涛(阜外华中心血管病医院),程应樟(南昌大学第二附属医院),解卫平(南京医科大学第一附属医院),潘磊(首都医科大学附属北京世纪坛医院)
秘书组成员(按姓氏笔画排序):陈剑飞(陆军军医大学新桥医院),徐希奇(中国医学科学院阜外医院),蒋鑫(中国医学科学院阜外医院)
无

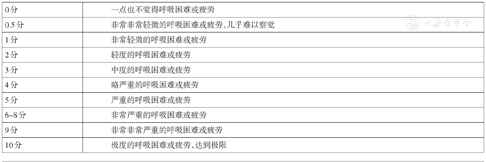
| 0分 | 一点也不觉得呼吸困难或疲劳 |
| 0.5分 | 非常非常轻微的呼吸困难或疲劳,几乎难以察觉 |
| 1分 | 非常轻微的呼吸困难或疲劳 |
| 2分 | 轻度的呼吸困难或疲劳 |
| 3分 | 中度的呼吸困难或疲劳 |
| 4分 | 略严重的呼吸困难或疲劳 |
| 5分 | 严重的呼吸困难或疲劳 |
| 6~8分 | 非常严重的呼吸困难或疲劳 |
| 9分 | 非常非常严重的呼吸困难或疲劳 |
| 10分 | 极度的呼吸困难或疲劳,达到极限 |

























