
Andersen-Tawil综合征1型(ATS1)是编码Kir2.1离子通道蛋白的KCNJ2基因发生突变的罕见的遗传性疾病,临床表现为周期性麻痹、QT/QTc间期延长、室性心律失常和发育畸形三联征,可无明显心脏相关表现,有持续性室性心动过速合并晕厥发作史者为心原性猝死的高危人群。本文报道1例典型的ATS1患者,频繁发作室性心动过速及晕厥,置入心脏复律除颤器治疗后出院,随访1个月预后良好。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
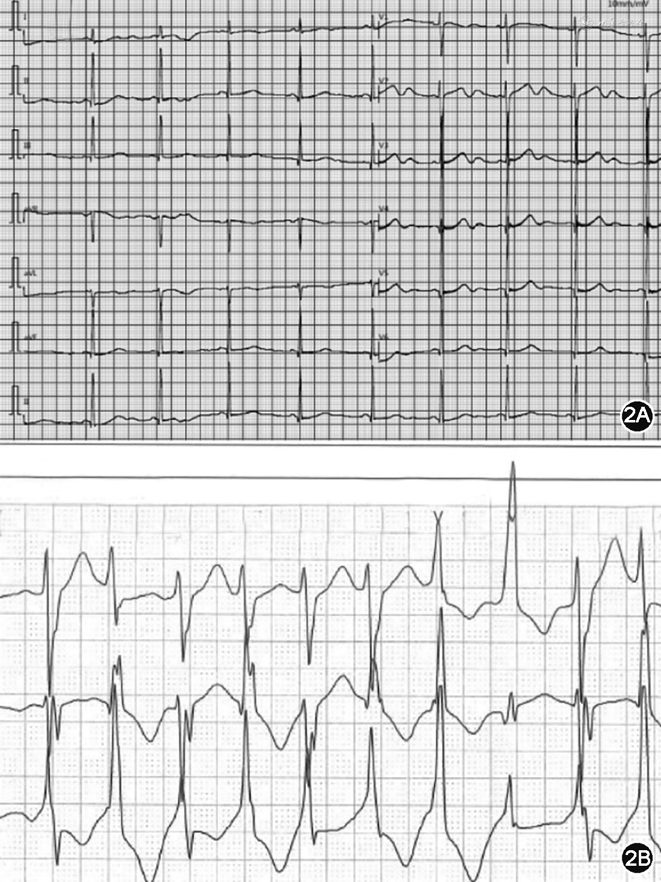
患者女,19岁,因“双下肢无力6年余,反复晕厥4年,再发6 d”于2021年3月17日收住浙江大学附属第二医院心内科。2014年始患者无明显诱因出现双下肢无力,主要为双小腿部,走路或上楼梯时加重,持续3~7 d可自行缓解。当时查体:双下肢肌力5-级,四肢腱反射减弱,双下肢关节位置觉稍差。血液检查:血钾2.97 mmol/L。心电图:窦性心律,频发室性早搏,QT间期延长,可见T-U融合。心脏超声未见明显异常。动态心电图可见频发多源性室性早搏,短阵双向室性心动过速(室速)。肌电图提示周期性麻痹。患者全外显子基因测序结果提示KCNJ2基因突变,突变位点类型p.G215D。临床诊断为Andersen-Tawil综合征(Andersen-Tawil Syndrome,ATS)1型(ATS1),阵发性室速。患者服用普萘洛尔和阿替洛尔均不能耐受,表现为咽喉部极度不适感,长期服用醋甲唑胺、倍他乐克和辅酶Q10。患者于4年前行走时再发心悸、头晕,随即意识丧失,2~3 min后自行恢复意识,无大小便失禁,此后晕厥反复发作3次;6 d前再次发作,于浙江大学附属第二医院住院。既往病史无特殊,父母非近亲结婚,父母及其姐姐基因检测均呈阴性。入院查体:脉率75 次/min,血压107/52 mmHg(1 mm Hg=0.133 kPa)。神志清,精神可,宽眼距,低耳廓,宽鼻,小下颌(图1A),双手第5小指弯曲(图1B)。心率75 次/min,心律不齐,心音中等,未闻及明显杂音及心包摩擦音;心电图提示窦性心律,左心室高电压,QT间期延长(QT/QTc间期448/504 ms),U波显著(图2A)。心脏超声心动图和心脏增强磁共振检查未见明显异常。心电监护见频发多源性室性早搏,室性二联律,非持续性室速,多数呈双向性室速,QT间期延长,U波宽大(图2B)。于2021年3月23日置入置入式心脏复律除颤器(implantable cardioverter defibrillator,ICD;Evera DDBC3D1,美敦力公司,美国)。术后程控以100 次/min起搏,心电图提示心房起搏心律,QT/QTc间期360/467 ms,心电监护未见室性心律失常,于2021年3月25日出院。术后1个月随访,心电图和ICD程控未见室速再发。
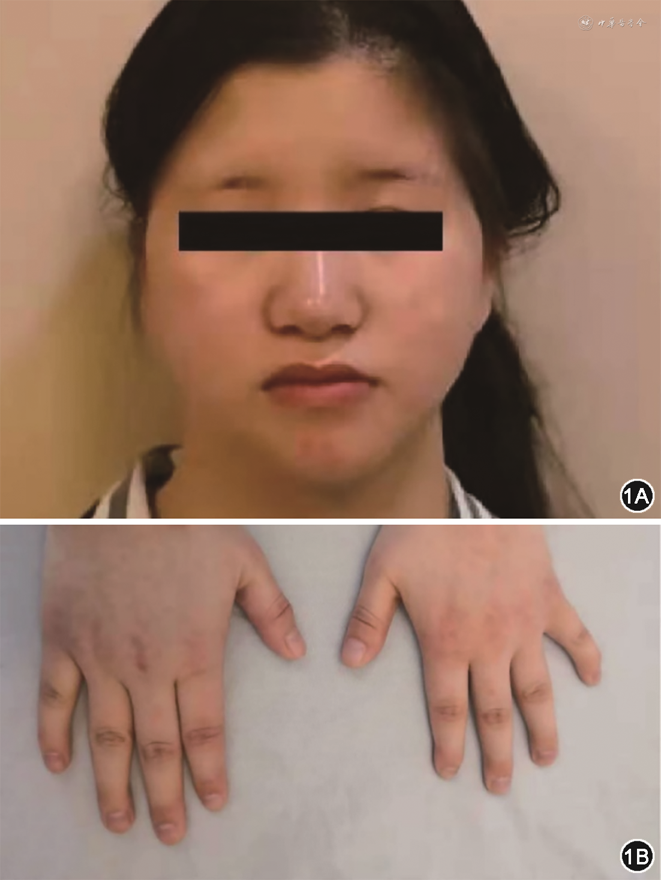
ATS是常染色体显性遗传病,发病率约百万分之一,临床表现为典型的周期性麻痹、QT/QTc间期延长、室性心律失常和发育畸形三联征[1],在分型中属于长QT间期综合征(long QT syndrome,LQTS)7型;根据基因突变类型又可分为2种亚型,ATS1约占60%,与KCNJ2基因突变相关;2型(ATS2)约占40%,可能与KCNJ5基因突变相关。KCNJ2基因位于17号染色体q23区域,编码离子通道蛋白Kir2.1[2]。Kir2.1蛋白主要表达于心脏、大脑以及骨骼肌,尤其是心肌细胞,其强大的内向整流钾电流(Ik1)作用有助于稳定细胞膜的静息电位以及调节复极晚期钾电流。KCNJ2基因突变时Ik1电流减少,导致心肌细胞静息膜电位部分去极化,动作电位时程和QT间期延长,可能诱发长QT相关的恶性室性心律失常。ATS基因常外显不完全,临床表型异质性大,约80%的患者具备两种及以上表现,具备完整典型三联征的患者不多见。临床诊断有2个标准:(1)具备3种典型特征中的2种:①周期性麻痹;②QTc间期延长和室性心律失常;③ATS的特殊面容:低位耳,宽眼距,小下颌,第5指(趾)弯曲畸形,并指;(2)具备上述3种典型特征之一,同时有一位家庭成员满足第(1)条标准。当患者临床表现符合上述标准之一时应疑诊为ATS;如果同时基因检测到KCNJ2基因突变可确诊为ATS[3]。
LQTS是最常见的遗传性心律失常,也是无器质性心脏病的年轻人猝死的常见病因之一。QT间期延长一度被认为是ATS患者心电图的主要特征性表现。早在1994年,Tawil等[1]就报道了来自3个家系的4位患者的心电图表现,多为QTc延长和各种室性心律失常,通过家系基因图谱分析,认为有发生恶性心律失常和猝死的致命性风险,并将其归类为LQT7。Zhang等[4]认为,ATS1患者心电图U波的出现影响QT/QTc测量的准确性,去除U波,ATS1患者的QT间期几乎均为正常,建议将ATS1与LQTS区分开。2020年Mazzanti等[5]发表了大规模的ATS1患者注册研究,共入选全球9个国家23个中心57个家系118例患者,进行全面的数据采集与分析后发现QT间期延长并不是诊断的必要条件,而显著U波可作为该疾病的一个特征性表现。因此ATS在LQTS分类中的意义还有一定争议。
相较其他类型的LQTS,ATS患者总体预后较好。ATS的处理目前尚依据经验性治疗,β受体阻滞剂单独使用或联合Ⅰc类抗心律失常药物(比如氟卡恩)效果良好[6, 7]。然而,2020年Mazzanti等[5]的注册研究表明,ATS患者恶性心律失常事件发生率较高,心律失常的危险因素包括不明原因的晕厥以及持续性的室速,β受体阻滞剂单用/联合Ⅰc类抗心律失常药物则不能降低恶性心律失常事件的发生率,胺碘酮可升高死亡率,采用ICD进行一级或二级预防均可在一定程度上降低心原性猝死发生率。
本文报道患者少年起病,有典型的临床三联征,临床及基因检测结果可确诊为ATS1。长期服用β受体阻滞剂后仍反复发作晕厥,心电图可见各种室性心律失常和宽大U波,QT/QTc间期明显延长,ICD置入后可有预防心脏性猝死的效果,但ICD基础上的药物治疗还需要进一步观察研究。
所有作者声明无利益冲突

























