
内耳畸形可分为迷路未发育、初始听泡、耳蜗未发育、共同腔畸形、耳蜗发育不全、不完全分隔型、大前庭导水管、前庭半规管畸形、蜗神经孔异常、前庭蜗神经异常。不完全分隔Ⅱ型和大前庭导水管与SLC26A4基因有关,不完全分隔Ⅲ型为一些X-连锁耳聋的表现。耳蜗发育不全可以与一些综合征性耳聋有关。对内耳畸形的完整评估应包括利用MRI对前庭蜗神经的评估。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
经全国继续医学教育委员会批准,本刊开设继教专栏,每年从第1期至第10期共刊发10篇继教文章,文后附5道单选题,读者阅读后可扫描标签二维码答题,每篇可免费获得Ⅱ类继教学分0.5分,全年最多可获5分。
内耳畸形是引起先天性感音神经性聋的常见原因。影像学检查是诊断内耳畸形的主要手段。目前内耳畸形的影像学分类已逐步完善,对其认识也逐步加深。本文中简要介绍内耳畸形的最新影像学分类及其研究进展。
内耳包埋在颞骨岩部的骨质内,位于鼓室和内耳道底之间,由骨性管道(骨迷路)和其深面的膜性管道(膜迷路)组成[1]。骨迷路是致密骨质围成的一系列弯曲小管和腔隙,分为3个部分:耳蜗、前庭和骨半规管。耳蜗位于内耳的前内侧,形似蜗壳;前庭位于中间,近似椭圆形;骨半规管由3个彼此相互垂直的“C”形骨管组成,分别称为上半规管、外半规管和后半规管,开口于前庭。膜迷路套在骨迷路内,是由上皮和结缔组织构成的膜性小管和囊,具有位、听感受器,为封闭的管道系统。膜迷路可分为蜗管、椭圆囊、球囊和膜半规管。蜗管套在骨螺旋管内;椭圆囊、球囊位于前庭的深面;膜半规管与骨半规管形态一致。膜迷路内充满内淋巴液,膜迷路与骨迷路壁之间充满外淋巴液,内、外淋巴液不相通。耳蜗底及前庭内侧壁朝向内听道底,有蜗神经和前庭神经的分支通过。此外,面神经也通过内听道底。
内耳主要由头部外胚层演变而来[1]。在胚胎第4周时,菱脑两侧的表面外胚层增厚形成听板,继之向下方间充质内陷,边缘向中央靠拢、闭合,形成耳泡(亦称听泡)。耳泡及其周围的间充质逐渐分化为内耳膜迷路。胚胎约6周时,膜半规管开始发生,上半规管最早,其次为后半规管,外半规管最晚;7~9周时,蜗管延长并开始卷曲;约3个月时,膜迷路周围的间充质分化成软骨囊,包绕膜迷路;约5个月时,软骨囊骨化成骨迷路[2]。内耳发育过程中出现停滞或变异就会导致内耳畸形。
内耳畸形可发生于骨迷路和膜迷路的任何部分,其中约80%为膜迷路畸形,约20%为膜迷路伴骨迷路畸形[3]。目前的常规CT及MRI只能检出骨迷路畸形。
高分辨CT(high resolution CT,HRCT)是内耳畸形的首选检查方法,最薄层厚可达0.5~0.6 mm。锥形束CT具有更高的空间分辨率(层厚可达0.1~0.3 mm)和更低的辐射剂量,在耳科影像方面应用越来越受到重视。通过多平面重建(multiplanar reconstruction,MPR)方法重建出标准层面并进行测量,有助于更准确地诊断内耳畸形以及术前评估[4]。
MRI的软组织分辨率高,可以观察内耳的形态及信号改变,还可显示前庭蜗神经发育异常等情况。内耳水成像能够重建出含水迷路腔的三维图像,更加立体地显示各种内耳畸形的形态。但值得注意的是,水成像中内淋巴液、外淋巴液均呈高信号,不能单独显示内淋巴液,故显示出的形态实际为骨迷路的形态。内耳水成像可以通过MPR对前庭蜗神经进行观察,而直接采集的垂直于内听道的斜矢状面图像对于前庭蜗神经各分支及面神经的显示更加清晰。
Jackler等[3]在1987年提出了根据胚胎发育过程的内耳畸形分类方法,但当时的影像检查手段为体层摄影。在此基础上,Sennaroglu等[5]在2002年提出了基于CT的内耳畸形分类法。在其最新的2017年版本中,进一步增加了MRI对于前庭蜗神经的评估[6]。
1.迷路未发育(complete labyrinthine aplasia,CLA):也称为Michel畸形,影像表现为内耳结构完全缺失,但骨迷路包囊之密质骨可以存在,可伴有颞部岩部发育不良。CLA可分为3个亚型:CLA伴岩锥发育不良或未发育(图1)、CLA不伴骨迷路包囊、CLA伴骨迷路包囊。
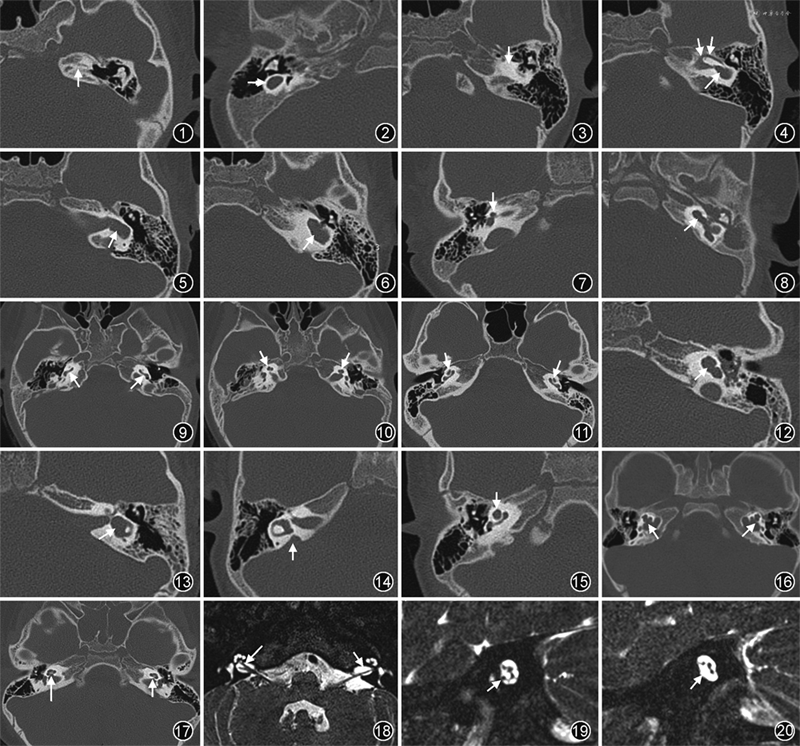
2.初始听泡:内耳表现为数毫米的小囊状,无内听道(图2),可以存在部分半规管结构。
3.耳蜗未发育:表现为耳蜗缺失(图3),可分为伴正常前庭半规管和伴前庭扩大(图4)两个亚型。后者需与共同腔畸形相鉴别,但有时很难区分。
4.共同腔畸形:前庭和耳蜗呈单一或融合腔,内听道通常进入其中部,可伴有部分发育的半规管(图5,6)。
5.耳蜗发育不全(cochlear hypoplasia,CH):耳蜗可以与前庭相区分,且耳蜗的尺寸小于正常,占内耳畸形的15%~23%[7]。CH影像表现多样,也常伴有前庭半规管畸形、蜗神经孔及内听道异常等。CH可分为4型:CH-Ⅰ,芽状耳蜗(图7);CH-Ⅱ,囊性耳蜗发育不全(图8);CH-Ⅲ,耳蜗圈数少于2圈(图9,10);CH-Ⅳ,耳蜗尖中旋发育不全(图11)。耳蜗宽度和高度的测量有助于提高诊断CH的敏感性[4]。
6.不完全分隔(incomplete partitian,IP):可分为3个亚型。IP-Ⅰ,约占内耳畸形的20%,又称为囊性耳蜗前庭畸形,耳蜗呈囊性外观,蜗轴及耳蜗分隔完全缺失,常伴有扩张的前庭,但耳蜗和前庭可以相区分,耳蜗的大小与正常相仿(图12,13)。IP-Ⅱ,即经典Mondini畸形,是最常见的耳蜗畸形,包括3个典型征象:蜗轴尖部缺陷,导致耳蜗尖中旋融合呈囊样;前庭轻度扩大;前庭导水管扩大(图14,15)。IP-Ⅲ,占内耳畸形的0.9%~2.0%,其特征表现为内听道球样扩张、蜗轴完全缺失,但耳蜗分旋存在(图16),可累及整个骨迷路包囊,包括前庭、半规管、前庭窗等[8]。
7.大前庭导水管(enlarged vestibular aqueduct,EVA):仅表现为前庭导水管扩大,而耳蜗、前庭半规管正常。诊断EVA的传统标准(Valvassori标准)为前庭导水管中点处宽度>1.5 mm;也有学者推荐采用Cincinnati标准,即前庭导水管中点处≥1 mm或后部开口处≥2 mm[9, 10]。
8.前庭半规管畸形:可表现为前庭扩大、半规管短小、狭窄、增宽,以及半规管缺失等。单独的前庭或半规管畸形很常见,其他内耳畸形也常伴有不同程度的前庭半规管畸形。
9.蜗神经孔异常:正常蜗神经孔的宽度为1.4~2.5 mm,其宽度<1.4 mm可考虑为蜗神经孔狭窄,也可表现为蜗神经孔闭锁(图17),可伴有或不伴有内听道狭窄。
10.前庭蜗神经异常:通常,HRCT可以对内耳畸形进行正确的诊断和分类,但完整的评估应包括MRI对于前庭蜗神经的评估,尤其是对蜗神经的评估,可表现为神经发育不良或缺失(图18~20)。在单侧重度及极重度感音神经性聋的儿童患者中,蜗神经发育不良或缺失的发生率可达48%[11]。
内耳畸形可以由基因异常所导致,可以为综合征性或非综合征性。与基因相关的内耳畸形常表现为双侧畸形,且双侧形态大致对称。
EVA及IP-Ⅱ可见于非综合征性耳聋患者中,也可发生在耳聋-甲状腺肿综合征(Pendred综合征)患者中,均由SLC26A4基因突变引起,表现为常染色体隐性遗传[12]。有研究表明,中国人群EVA及IP-Ⅱ患者中,92%为SLC26A4双等位基因突变,6%为单等位基因突变[13]。
IP-Ⅲ型为X-连锁耳聋2型(DFNX2)的典型影像学表现,也是最常见的一种X-连锁非综合征性耳聋,与POU3F4基因有关[14]。部分病例可伴有下丘脑畸形或错构瘤表现[15, 16]。也有报道COL4A6基因突变导致的X-连锁耳聋也呈IP-Ⅲ型表现[17]。
CH与前庭半规管畸形常与一些综合征性耳聋相关。如CHARGE综合征典型内耳畸形表现为半规管未发育伴前庭发育不良,伴有CH、蜗神经孔狭窄或闭锁,可伴有外中耳畸形[7]。鳃-耳-肾综合征表现耳蜗尖中旋发育不全、EVA、面神经管迷路段内移、外中耳畸形等[7]。Waardenburg综合征可表现为后半规管缺失、CH、EVA等[7]。
中华医学会放射学分会头颈学组对本栏目给予大力支持
所有作者声明无利益冲突
1.评估蜗神经宜采用哪种检查技术()
A.高分辨CT
B.锥形束CT
C.MR水成像
D.MR增强
2.当前的影像学检查手段不能发现下列哪一类畸形()
A.骨迷路畸形
B.膜迷路畸形
C.前庭蜗神经发育异常
D.面神经走行异常
3.关于内耳畸形,下列哪种说法是错误的()
A.耳蜗畸形一般不伴有前庭半规管畸形
B.耳蜗发育不全表现多样,共同点为耳蜗与前庭可以区分,耳蜗尺寸小于正常
C.不完全分隔畸形中耳蜗的外部尺寸与正常类似
D.耳蜗未发育伴前庭扩大有时难以与共同腔畸形相鉴别
4.关于大前庭导水管,下列哪种说法是错误的()
A.传统的Valvassori诊断标准为前庭导水管中点处宽度>1.5 mm
B.Cincinnati诊断标准为前庭导水管中点处≥1 mm或后部开口处≥2 mm
C.大前庭导水管不会合并其他内耳畸形
D.大前庭导水管及不完全分隔Ⅱ型可以与SLC26A4基因突变或Pendred综合征有关
5.哪一种内耳畸形在人工耳蜗植入时最常发生脑脊液喷涌()
A.共同腔畸形
B.耳蜗发育不全
C.不完全分隔Ⅰ型
D.不完全分隔Ⅲ型

























